This page assumes you have already used the structural-dependence overview or
one of the worked tutorials (animal-models,
phylogenetic-models, spatial-models,
relmat-known-matrices) to choose a route; it collects the
current fitted ladder across all of them in one place. Structural
dependence means a random effect has a known dependence structure
instead of independent group levels. Read the grammar in biological
order: animal models for pedigree or additive relatedness, phylogenetic
models for tree-structured relatedness, spatial models for coordinate or
mesh structure, combined phylogenetic-plus-spatial models when both
sources matter, and relmat() for other known relatedness or
precision matrices.
The current fitted drmTMB paths are a subset of that
ladder. For Gaussian models, phylo(), coordinate
spatial(), animal(), and relmat()
each fit one univariate mu intercept and one numeric
mu slope. These one-slope paths estimate independent
structured intercept and slope fields with separate SDs. Those Gaussian
mu terms do not themselves establish multiple or labelled
slopes, intercept-slope correlations, or structured residual
rho12. Separate exact q1 sigma and
non-Gaussian gates are fitted for the providers and families named in
the route table below. A first bivariate path also fits matching
intercept-only phylo() terms in mu1 and
mu2, estimating the two phylogenetic location SDs and their
mean-mean correlation. Matching labelled phylo() terms
across mu1, mu2, sigma1, and
sigma2 now fit the first constant bivariate phylogenetic
location-scale block. The first predictor-dependent phylogenetic
correlation regression is also fitted for the q=2
mu1-mu2 location-location row. The first
coordinate-based spatial paths,
spatial(1 | site, coords = coords) and
spatial(1 + x | site, coords = coords), are fitted for
univariate Gaussian location models. Matching labelled
spatial(1 | p | site, coords = coords) terms in bivariate
mu1 and mu2 fit the first coordinate-spatial
mean-mean correlation, reported by
corpairs(level = "spatial"); matching labelled spatial
terms across mu1, mu2, sigma1,
and sigma2 fit the first constant coordinate-spatial q=4
location-scale block. animal() and relmat()
fit the same first Gaussian intercept and one-slope mu
pattern when supplied with a pedigree, precomputed relationship, or
precision matrix, such as
animal(1 | id, pedigree = pedigree),
animal(1 + x | id, Ainv = Ainv), or
relmat(1 + x | id, Q = Q). Artifact routing is narrower
than fitted syntax. The one-slope routes have manual Phase 18 artifact
tasks named phylo_mu_slope, spatial_mu_slope,
animal_mu_slope, and relmat_mu_slope. These
tasks are opt-in, excluded from task = "all", and are not
recovery, coverage, or power evidence. Mesh/SPDE spatial fields,
combined phylogenetic-plus-spatial location models, additional multiple
or labelled spatial-slope layouts outside the exact fitted ledger cells,
spatial slope correlations, non-Gaussian spatial effects outside the
exact ordinary Poisson/NB2 q1 spatial mu
intercept-plus-one-slope, recovery-grade NB2 q1 spatial
sigma, Student-t spatial mu, Poisson spatial
zi, fixed-zi Poisson spatial mu,
and fixed-zi NB2 spatial mu gates, and
predictor-dependent spatial correlations remain planned extensions. The
q1 spatial sigma one-slope route has point-fit/extractor
evidence, but its interval gate remains blocked.
The scientific questions for the structural ladder are simple:
Do predictors explain trait means after accounting for resemblance among relatives, species, or nearby sites? Does a response still have smooth coordinate-based site deviations, or does a predictor’s slope vary smoothly across sites?
For animal models, individual or animal
indexes rows in the data. A small
id/dam/sire pedigree or a
precomputed additive relatedness or inverse-relatedness matrix supplies
expected genetic similarity. For phylogenetic models,
species indexes the rows in the data and tree
supplies the relatedness among those species. For spatial models,
site indexes the rows in the data and coords
supplies one coordinate pair per site, or one row per observation when
coordinates are constant within site. In the fitted animal,
phylogenetic, spatial, and relmat() paths, the structured
term adds Gaussian coefficients to the location parameter
mu. The intercept path adds an individual, species, site,
or matrix-indexed deviation in mean response. The one-slope paths let
the effect of one numeric predictor vary across the same structured
levels.
Throughout this article, Normal(a, b) and
MVN(a, B) use variance or covariance as the second
argument.
In this article, a location parameter such as mu
describes the expected trait value, a scale parameter such as
sigma describes residual or random-effect variability, a
shape parameter such as nu describes a family-specific
feature such as tail weight or skewness, and a coscale parameter models
residual correlation, such as bivariate rho12. Structured
animal, phylogenetic, and spatial effects are not residual coscale terms
unless the fitted model explicitly includes a residual
rho12 formula.
Reader route: animal, phylo, spatial, phylo+spatial, relmat
Read this page as a five-step structural-dependence ladder. Step 1
asks whether individuals resemble each other because of pedigree or
additive relatedness. Step 2 asks whether related species remain similar
after fixed effects. Step 3 asks whether nearby sites remain similar
after fixed effects. Step 4 asks whether both species relatedness and
site coordinates are needed in the same model. Step 5 is the lower-level
relmat() route for known dependence matrices that do not
belong to the named animal, phylogenetic, or spatial routes. In the
current package, Steps 1, 2, 3, and 5 have fitted Gaussian paths plus
exact row-specific non-Gaussian gates; Step 4 is documented planned
syntax.
For a one-response phylogenetic-plus-spatial endpoint such as
heat_tolerance, the conceptual full model is:
Here is the observed trait, for example heat tolerance for individual or species-site observation . The vector contains location predictors such as habitat temperature or body mass, and are their average effects on the expected trait value. The term is the phylogenetic deviation for the species attached to observation , is the phylogenetic covariance implied by the tree, and is the SD of tree-structured location deviations. The term is the spatial deviation for the sampling site, is the coordinate-based spatial covariance, and is the SD of spatial location deviations. The vector contains residual-scale predictors and are their effects on log residual SD.
The animal-model analogue replaces the species term with an
individual additive effect, for example
.
That is the meaning of
animal(1 | individual, pedigree = pedigree),
animal(1 | individual, A = A), or
animal(1 | individual, Ainv = Ainv). The pedigree route
builds a dense additive relationship matrix for this first Gaussian
location path; sparse large-pedigree precision construction remains
planned.
Read the current route status this way:
| Route | Current status | Use when | Syntax to read |
|---|---|---|---|
| 1. Animal | Implemented for the documented Gaussian routes plus exact ordinary
Poisson/NB2 q1 mu intercept-plus-one-slope, NB2 q1
sigma, and beta animal gates at their recorded tiers;
sparse large-pedigree construction remains planned |
Individuals have known additive relatedness from a pedigree or relationship matrix, and the biological question is an animal model or quantitative-genetic random effect. |
animal(1 | individual, pedigree = pedigree),
animal(1 + x | individual, Ainv = Ainv), or matching
labelled Gaussian terms; consult the live ledger before using a
non-Gaussian animal gate |
| 2. Phylo | Implemented for the documented Gaussian routes plus exact ordinary
Poisson/NB2 q1 mu intercept-plus-one-slope, NB2 q1
sigma, Student-t q1 nu, cumulative-logit q1
mu, and recovery-grade Gamma/lognormal q1 mu
gates |
Species share evolutionary history, and related species may have similar trait means, trait variances, or cross-trait latent correlations. |
phylo(1 | species, tree = tree),
phylo(1 + x | species, tree = tree), labelled Gaussian
blocks, or one exact non-Gaussian gate at its recorded tier |
| 3. Spatial | Implemented for the documented Gaussian routes plus exact ordinary
Poisson/NB2 q1 mu intercept-plus-one-slope, NB2 q1
sigma, Student-t q1 mu, Poisson q1
zi, fixed-zi Poisson q1 mu, and
fixed-zi NB2 q1 mu gates |
Nearby sampling sites may share smooth deviations in the expected
response, in one numeric slope such as depth, or in paired
response means. |
spatial(1 | site, coords = coords),
spatial(1 + depth | site, coords = coords), matching
labelled Gaussian terms, or one exact non-Gaussian gate at its recorded
tier |
| 4. Phylo + spatial | Planned, not fitted yet | The same response could be structured by both species relatedness and site coordinates. | Fit separate phylogenetic and spatial sensitivity models for now;
simultaneous phylo() plus spatial() in
mu is rejected until the joint model has identifiability
checks. |
5. relmat()
|
Implemented for the documented Gaussian routes plus exact ordinary
Poisson/NB2 q1 mu intercept-plus-one-slope, NB2 q1
sigma, recovery-grade Gamma/lognormal q1 mu,
and truncated-NB2 q1 hu gates |
A known covariance or precision matrix does not naturally belong to animal, phylogenetic, or spatial syntax. |
relmat(1 | id, K = K),
relmat(1 + x | id, Q = Q), matching labelled Gaussian
terms, or one exact non-Gaussian gate at its recorded tier |
That status boundary is deliberate. A combined model like this is scientifically natural:
# Planned, not fitted yet:
heat_tolerance ~ climate +
phylo(1 | species, tree = tree) +
spatial(1 | site, coords = coords)It requires careful checks that the tree-structured and
coordinate-structured SDs are both identifiable before
drmTMB treats the syntax as runnable.
For animal and relmat() models, use the known-matrix
slices when the matrix is already available and the target is a Gaussian
mu or sigma intercept, one numeric Gaussian
mu slope, a two-response q=2 location covariance, or a
constant q=4 location-scale block. If the question is repeatability
without known relatedness, fit an ordinary (1 | individual)
or (1 | line) Gaussian model and report that it ignores
pedigree or matrix structure. If the question is species shared
ancestry, use the fitted phylo() route. If the question is
site structure, use the fitted coordinate-spatial route. If the matrix
is sampling covariance among effect-size estimates, use
meta_V(V = V), not relmat();
relmat() is for latent random-effect relatedness or
precision matrices.
| Current question | Syntax | Fitted action now |
|---|---|---|
| Animal-model additive individual deviations from a pedigree | animal(1 | individual, pedigree = pedigree) |
Use the fitted univariate Gaussian mu first slice;
inspect check_drm() and profile_targets()
before interpreting the SD. |
| Additive genetic variance from a precomputed relationship matrix |
animal(1 | individual, A = A) or
animal(1 | individual, Ainv = Ainv)
|
Use the fitted univariate Gaussian mu first slice;
inspect check_drm() and profile_targets()
before interpreting the SD. |
| Experimental-line, genomic, or laboratory relatedness that is not a pedigree, tree, or coordinate surface |
relmat(1 | line, K = K) or
relmat(1 | line, Q = Q)
|
Use the fitted univariate Gaussian mu first slice, or
switch to phylo() / spatial() when the known
structure belongs to those named routes. |
| Known sampling covariance among effect-size estimates | meta_V(V = V) |
Use the fitted Gaussian meta-analysis route; this is observation covariance, not a latent relatedness random effect. |
Animal model with a known-matrix first slice
An applied animal-model question might be: after accounting for age and sex, is there additive individual-level variation in body size that follows a known pedigree? The fitted first slice uses a precomputed additive relationship or inverse relationship matrix:
fit_animal <- drmTMB(
drm_formula(
body_size ~ age + sex + animal(1 | individual, Ainv = Ainv),
sigma ~ sex
),
family = gaussian(),
data = growth
)That animal effect puts the individual-level location deviations in a
precomputed additive relationship matrix. It is not a residual
rho12 coscale model, and it does not use
meta_V(V = V), which is reserved for known sampling
covariance among observations or effect-size estimates.
animal_example <- simulate_animal_marker_data(seed = 1)
growth <- animal_example$data
Ainv <- animal_example$Ainv
fit_animal_known <- drmTMB(
drm_formula(
body_size ~ age + sex + animal(1 | individual, Ainv = Ainv),
sigma ~ sex
),
family = gaussian(),
data = growth
)
check_drm(fit_animal_known)
#> <drm_check: 14 checks>
#> ok: 14; notes: 0; warnings: 0; errors: 0
#> check status
#> optimizer_convergence ok
#> optimizer_budget ok
#> finite_objective ok
#> logsigma_clamp_active ok
#> fixed_gradient ok
#> sdreport_status ok
#> hessian_positive_definite ok
#> standard_errors_finite ok
#> standard_errors_inflated ok
#> dropped_rows ok
#> positive_scale ok
#> random_effect_sd_boundary ok
#> fixed_effect_design_size ok
#> animal_mu_diagnostics ok
#> value
#> 0
#> iterations=30; function=41; gradient=31
#> 111.5
#> <NA>
#> max=0.00005840; component=beta_sigma[1]
#> ok
#> TRUE
#> range=[0.06910,0.1271]
#> n_inflated=0; max_se=0.1271; median_se=0.1105
#> nobs=160; dropped=0
#> min=0.3969
#> min=0.3700; boundary=0.0001000; term=mu.animal(1 | individual)
#> total_mb=0.02820; max_cols=3; largest=mu; largest_class=matrix; largest_density=0.8333
#> group=individual; n_nodes=40; n_observed=40; min_group_n=4; matrix_type=precision; structured_sd=0.3700; sd_ratio=0.9170
#> message
#> nlminb convergence code is 0.
#> Optimizer evaluation counts recorded; no eval.max or iter.max control was supplied.
#> Objective and log-likelihood are finite.
#> The log(sigma) clamp is not active at the optimum.
#> Maximum absolute fixed gradient is <= 0.001; largest component is beta_sigma[1].
#> TMB::sdreport() completed successfully.
#> sdreport reports a positive-definite Hessian.
#> All fixed-effect standard errors are finite.
#> No fixed-effect standard error is inflated relative to the others.
#> No rows were dropped by model-frame or known-covariance filtering.
#> All fitted scale values are finite and positive.
#> All fitted random-effect standard deviations are finite, positive, and above the requested lower-boundary warning threshold.
#> Dense fixed-effect design matrices are modest for this fit.
#> The animal-model random intercept has replicated observed levels and a non-negligible fitted SD relative to residual scale.
summary(fit_animal_known)
#> <summary.drmTMB>
#> estimator: ML
#> estimate std_error
#> mu:(Intercept) 2.09026748 0.12452429
#> mu:age 0.55224388 0.06910001
#> mu:sexmale 0.29853712 0.11045158
#> sigma:(Intercept) -0.92404183 0.08895052
#> sigma:sexmale 0.03257437 0.12713997
#> Distributional, random-effect, scale, and correlation parameters:
#> component dpar term
#> sd:mu:animal(1 | individual) random-effect-sd mu animal(1 | individual)
#> fitted:sigma distributional-scale sigma fitted range
#> estimate std_error minimum maximum scale
#> sd:mu:animal(1 | individual) 0.3700002 0.05265305 NA NA response
#> fitted:sigma 0.4034826 NA 0.3969115 0.4100536 response
#> logLik: -111.5
#> convergence: 0If two traits were measured on the same individuals, a matching label
such as p asks whether the additive deviations in the two
location predictors covary. That covariance is a latent animal-model
row; residual rho12 still describes within-observation
residual correlation after the known relatedness effect is accounted
for.
fit_animal_q2_known <- drmTMB(
drm_formula(
mu1 = body_size ~ age + sex +
animal(1 | p | individual, Ainv = Ainv),
mu2 = beak_length ~ age + sex +
animal(1 | p | individual, Ainv = Ainv),
sigma1 = ~ sex,
sigma2 = ~ sex,
rho12 = ~ 1
),
family = c(gaussian(), gaussian()),
data = growth
)
animal_q2_pairs <- corpairs(
fit_animal_q2_known,
level = "animal"
)
animal_q2_pairs
#> level group block from_dpar to_dpar from_coef to_coef
#> 1 animal individual p mu1 mu2 (Intercept) (Intercept)
#> from_response to_response class
#> 1 body_size beak_length mean-mean
#> parameter estimate min
#> 1 cor(mu1:(Intercept),mu2:(Intercept) | p | individual) 0.3657736 0.3657736
#> max n_values link_estimate link_min link_max modelled conf.status
#> 1 0.3657736 1 0.3835356 0.3835356 0.3835356 FALSE not_requested
#> interval_source
#> 1 not_available
animal_q2_checks <- check_drm(fit_animal_q2_known)
animal_q2_checks[grepl("animal|convergence|hessian", animal_q2_checks$check), ]
#> <drm_check: 4 checks>
#> ok: 4; notes: 0; warnings: 0; errors: 0
#> check status
#> optimizer_convergence ok
#> hessian_positive_definite ok
#> animal_mu_diagnostics ok
#> biv_animal_q2_covariance ok
#> value
#> 0
#> TRUE
#> group=individual; n_nodes=40; n_observed=40; min_group_n=4; matrix_type=precision; n_coef=2; min_structured_sd=0.2364; min_sd_ratio=0.6284
#> group=individual; rho_abs=0.3658; boundary=0.9800; n_levels=40; min_level_n=4; min_sd_ratio=0.6780
#> message
#> nlminb convergence code is 0.
#> sdreport reports a positive-definite Hessian.
#> The animal-model random intercept has replicated observed levels and a non-negligible fitted SD relative to residual scale.
#> Animal-model q2 location covariance has replicated levels, non-negligible fitted component SDs, and a latent correlation away from the boundary.
if (requireNamespace("ggplot2", quietly = TRUE)) {
animal_q2_display <- animal_q2_pairs
animal_q2_display$display_label <- "Animal\nmu1-mu2"
plot_corpairs(
animal_q2_display,
colour = "level",
label = "display_label",
facet = NULL
) +
ggplot2::scale_colour_manual(values = c("animal" = "#0072B2")) +
ggplot2::scale_fill_manual(values = c("animal" = "#0072B2")) +
structural_eye_theme() +
ggplot2::labs(
title = "Animal-model latent mean correlation",
subtitle = "Point estimate only; dotted line marks zero",
x = "Correlation estimate"
)
}
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's fill values.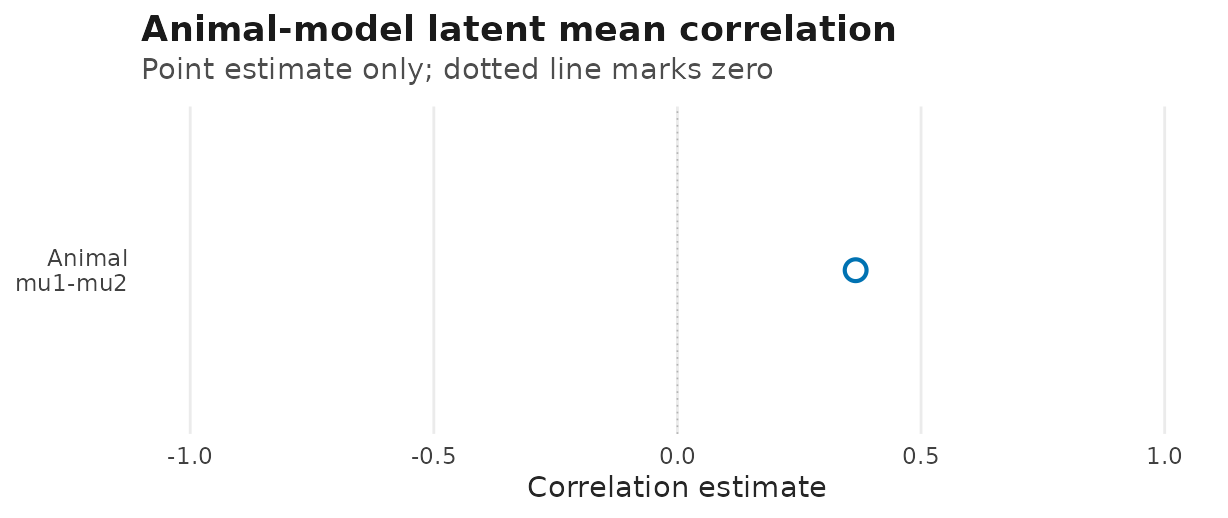
Animal-model q=2 location-location point estimate from
corpairs(); the dotted vertical line marks zero
correlation. Interval calibration for this intercept-only row is not yet
validated.
Use relmat() when the matrix is not naturally a
pedigree, tree, or spatial coordinate surface. Here Ginv is
a precision matrix for experimental breeding lines, so the fitted row is
a lower-level relatedness covariance rather than an animal-model
covariance.
Ginv <- animal_example$Ginv
fit_relmat_q2_known <- drmTMB(
drm_formula(
mu1 = body_size ~ age + sex +
relmat(1 | p | breeding_line, Q = Ginv),
mu2 = beak_length ~ age + sex +
relmat(1 | p | breeding_line, Q = Ginv),
sigma1 = ~ sex,
sigma2 = ~ sex,
rho12 = ~ 1
),
family = c(gaussian(), gaussian()),
data = growth
)
relmat_q2_pairs <- corpairs(
fit_relmat_q2_known,
level = "relmat"
)
relmat_q2_pairs
#> level group block from_dpar to_dpar from_coef to_coef
#> 1 relmat breeding_line p mu1 mu2 (Intercept) (Intercept)
#> from_response to_response class
#> 1 body_size beak_length mean-mean
#> parameter estimate min
#> 1 cor(mu1:(Intercept),mu2:(Intercept) | p | breeding_line) 0.4079354 0.4079354
#> max n_values link_estimate link_min link_max modelled conf.status
#> 1 0.4079354 1 0.4331324 0.4331324 0.4331324 FALSE not_requested
#> interval_source
#> 1 not_available
relmat_q2_targets <- profile_targets(fit_relmat_q2_known)
relmat_q2_targets[
grepl("relmat", relmat_q2_targets$parm),
c("parm", "target_type", "profile_ready")
]
#> parm
#> 13 sd:mu:mu1:relmat(1 | p | breeding_line)
#> 14 sd:mu:mu2:relmat(1 | p | breeding_line)
#> 15 cor:relmat:cor(mu1:(Intercept),mu2:(Intercept) | p | breeding_line)
#> target_type profile_ready
#> 13 direct TRUE
#> 14 direct TRUE
#> 15 direct TRUE
if (requireNamespace("ggplot2", quietly = TRUE)) {
relmat_q2_display <- relmat_q2_pairs
relmat_q2_display$display_label <- "relmat\nmu1-mu2"
plot_corpairs(
relmat_q2_display,
colour = "level",
label = "display_label",
facet = NULL
) +
ggplot2::scale_colour_manual(values = c("relmat" = "#6A6A6A")) +
ggplot2::scale_fill_manual(values = c("relmat" = "#6A6A6A")) +
structural_eye_theme() +
ggplot2::labs(
title = "Known-matrix latent mean correlation",
subtitle = "Point estimate only; dotted line marks zero",
x = "Correlation estimate"
)
}
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's fill values.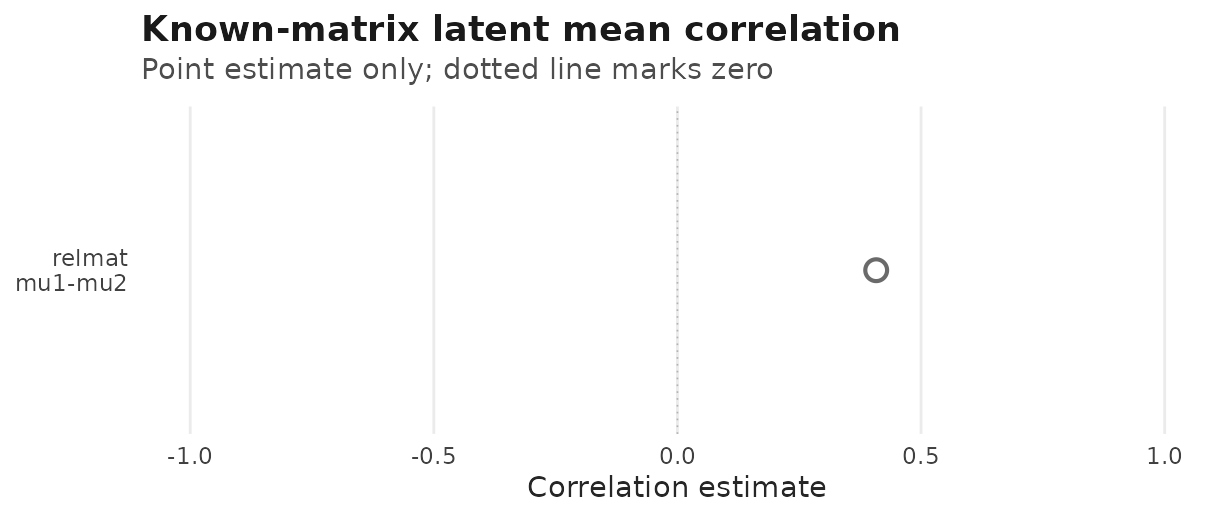
relmat() q=2 location-location point estimate from
corpairs(); the dotted vertical line marks zero
correlation. Interval calibration for this intercept-only row is not yet
validated.
When the relationship matrix is unavailable, fit an ordinary grouped Gaussian model as a sensitivity analysis and state the omitted structure:
fit_repeatability <- drmTMB(
drm_formula(
body_size ~ age + sex + (1 | individual),
sigma ~ sex
),
family = gaussian(),
data = growth
)
check_drm(fit_repeatability)
#> <drm_check: 14 checks>
#> ok: 14; notes: 0; warnings: 0; errors: 0
#> check status
#> optimizer_convergence ok
#> optimizer_budget ok
#> finite_objective ok
#> logsigma_clamp_active ok
#> fixed_gradient ok
#> sdreport_status ok
#> hessian_positive_definite ok
#> standard_errors_finite ok
#> standard_errors_inflated ok
#> dropped_rows ok
#> positive_scale ok
#> random_effect_sd_boundary ok
#> fixed_effect_design_size ok
#> mu_random_effect_replication ok
#> value
#> 0
#> iterations=20; function=30; gradient=21
#> 115.7
#> <NA>
#> max=0.0006233; component=beta_mu[2]
#> ok
#> TRUE
#> range=[0.07063,0.1466]
#> n_inflated=0; max_se=0.1466; median_se=0.1231
#> nobs=160; dropped=0
#> min=0.3998
#> min=0.4160; boundary=0.0001000; term=mu.(1 | individual)
#> total_mb=0.02820; max_cols=3; largest=mu; largest_class=matrix; largest_density=0.8333
#> (1 | individual)=4
#> message
#> nlminb convergence code is 0.
#> Optimizer evaluation counts recorded; no eval.max or iter.max control was supplied.
#> Objective and log-likelihood are finite.
#> The log(sigma) clamp is not active at the optimum.
#> Maximum absolute fixed gradient is <= 0.001; largest component is beta_mu[2].
#> TMB::sdreport() completed successfully.
#> sdreport reports a positive-definite Hessian.
#> All fixed-effect standard errors are finite.
#> No fixed-effect standard error is inflated relative to the others.
#> No rows were dropped by model-frame or known-covariance filtering.
#> All fitted scale values are finite and positive.
#> All fitted random-effect standard deviations are finite, positive, and above the requested lower-boundary warning threshold.
#> Dense fixed-effect design matrices are modest for this fit.
#> Every random-effect level has at least two fitted observations.That fallback asks whether repeated observations of the same
individual share a common location deviation. It does not estimate
additive genetic variance, does not use pedigree relatedness among
individuals, and cannot distinguish genetic resemblance from permanent
environmental or measurement effects. If the scientific claim requires
additive genetic variance, use the fitted animal(A/Ainv)
route once the matrix is ready; report the ordinary random-effect fit
only as a sensitivity check.
For matrix inputs, keep the representation explicit. A covariance
matrix such as A or K is the dense VCV form.
It is useful for small examples and parity checks, but it does not by
itself justify large-pedigree or large-matrix claims. A precision or
inverse relationship matrix such as Ainv or Q
is the preferred scalable route for larger examples. The first slice
validates row-name matching, determinant handling, positive
definiteness, diagnostics, and recovery tests, but speed claims still
need larger sparse-matrix evidence. This is the same distinction as the
implemented phylogenetic tree path: users pass tree = tree,
and drmTMB builds the sparse augmented A-inverse internally
rather than asking users to hand-write a dense tip VCV.
Parity target for animal, spatial, and relmat
The long-term target is not just to parse animal(),
spatial(), or relmat(). These routes should
eventually answer the same classes of questions as the fitted
phylogenetic route, whenever the corresponding dependence structure is
scientifically meaningful and identifiable. The table below is a roadmap
boundary, not a fitted-claim table.
| Layer | Fitted phylogenetic example | Parity target for animal, spatial, and relmat |
|---|---|---|
| Univariate location structure | phylo(1 | species, tree = tree) |
animal(1 | individual, pedigree = pedigree),
spatial(1 | site, coords = coords), and
relmat(1 | line, K = K) in mu
|
| Bivariate location-location correlation | Matching labelled phylo() terms in mu1 and
mu2, reported by corpairs()
|
Matching labelled spatial(), animal(), and
relmat() terms now fit the first q=2 slices when their
coordinate or known-matrix inputs are available |
| Location-scale q=4 block | Matching labelled phylo() terms across
mu1, mu2, sigma1, and
sigma2
|
Matching labelled all-four spatial, animal, and
relmat() blocks now fit constant q=4 location-scale
covariance |
| Predictor-dependent structured correlation | q=2 phylogenetic
corpair(..., level = "phylogenetic") ~ ecology
|
q=2 animal, spatial, and relmat corpair() regressions
only after constant q=2 fits are stable |
| Direct SD surfaces |
sd_phylo(), sd_phylo1(), and
sd_phylo2()
|
Source-specific direct-SD siblings after naming, matrix scale, and interpretation are clear |
Spatial is partway up this ladder:
spatial(1 | site, coords = coords) can enter Gaussian
mu, Gaussian sigma, or matching univariate
mu/sigma blocks; one independent
coordinate-spatial mu slope, the first bivariate
coordinate-spatial q=2 mu1/mu2 covariance, and
the constant q=4 location-scale covariance are fitted. Animal and
relmat() now have pedigree or known-matrix Gaussian
mu and sigma intercepts, matching univariate
mu/sigma correlations, q1 sigma
one-slope routes, matching labelled q=2 bivariate location covariance,
and matching all-four constant q=4 location-scale covariance. The exact
A-matrix animal and K/Q relmat sigma slopes are inference-ready with
caveats; the spatial sigma-slope interval gate remains blocked. Spatial
corpair(), mesh/SPDE fields, pedigree/Ainv bridge
marshalling, sparse large-pedigree construction, additional multiple or
labelled slope layouts outside the exact fitted ledger cells, and
animal/relmat() predictor-dependent corpair()
routes remain planned.
The focused animal, spatial, and relmat() pages cover
the current q=2 and constant q=4 routes. A fitted animal q=4 block looks
like:
fit_animal_q4 <- drmTMB(
drm_formula(
mu1 = body_size ~ age + sex +
animal(1 | p | individual, pedigree = pedigree),
mu2 = beak_length ~ age + sex +
animal(1 | p | individual, pedigree = pedigree),
sigma1 = ~ sex + animal(1 | p | individual, pedigree = pedigree),
sigma2 = ~ sex + animal(1 | p | individual, pedigree = pedigree),
rho12 = ~ 1
),
family = c(gaussian(), gaussian()),
data = growth
)
# Fitted spatial q=2 and q=4 sibling terms:
spatial(1 | p | site, coords = coords)
# Fitted relatedness q=2 and q=4 sibling terms:
relmat(1 | p | line, K = G)
relmat(1 | p | line, Q = Ginv)The next richer animal, spatial, and relmat() targets
are structured slopes and predictor-dependent structured correlations,
which remain planned.
Current implementation status
This page separates the fitted paths from the roadmap. The fitted
structural pieces are useful, but they are not yet the full animal,
phylo, spatial, phylo+spatial, and relmat()
location-scale-coscale ladder.
| Model piece | Status | What it means |
|---|---|---|
animal(1 | individual, pedigree = pedigree) |
Implemented first slice | A univariate Gaussian mu random intercept using a dense
additive relationship matrix built from id,
dam, and sire pedigree columns. Sparse
large-pedigree precision construction remains planned. |
animal(1 | individual, A = A) or
animal(1 | individual, Ainv = Ainv)
|
Implemented first slice | A univariate Gaussian mu random intercept using a
precomputed additive relatedness or inverse-relatedness matrix. Use
pedigree, A, or Ainv; keep
V for known sampling covariance in meta-analysis. |
animal(1 + x | individual, pedigree = pedigree) or
animal(1 + x | individual, Ainv = Ainv)
|
Implemented first one-slope slice | One trait has independent animal-model intercept and numeric slope fields in the location predictor. The two fields have separate SDs and do not estimate an intercept-slope correlation. |
Matching labelled
animal(1 | p | individual, pedigree = pedigree),
animal(1 | p | individual, A = A), or
animal(1 | p | individual, Ainv = Ainv) terms in bivariate
mu1 and mu2
|
Implemented first q=2 slice | Two Gaussian responses have correlated animal-model location
deviations from the same pedigree-derived or known matrix.
corpairs(level = "animal") reports this mean-mean row
separately from residual rho12. |
phylo(1 | species, tree = tree) in univariate
mu
|
Implemented | One trait has a phylogenetic random intercept in the location predictor. |
phylo(1 + x | species, tree = tree) in univariate
mu
|
Implemented first one-slope slice | One trait has independent phylogenetic intercept and numeric slope fields in the location predictor. The two fields share the same tree-derived covariance, have separate SDs, and do not estimate an intercept-slope correlation. |
Matching phylo(1 | species, tree = tree) terms in
bivariate mu1 and mu2
|
Implemented first slice | Two traits have correlated phylogenetic location deviations;
corpairs(level = "phylogenetic") reports the mean-mean row
separately from residual rho12. |
Matching labelled phylo(1 | p | species, tree = tree)
terms in bivariate mu1 and mu2
|
Implemented | The label is preserved in SD names, the fitted mean-mean
correlation, corpairs(block = "p"), and profile-target
names. |
Ordinary (1 | p | id) terms in all four bivariate
mu1, mu2, sigma1, and
sigma2 formulas |
Implemented first slice | One ordinary q=4 group block estimates six latent location-location, location-scale, and scale-scale correlations. |
Matching labelled phylo() terms in all four bivariate
mu1, mu2, sigma1, and
sigma2 formulas |
Implemented first slice | One constant q=4 phylogenetic block estimates four endpoint SDs and six latent correlations: one location-location, four location-scale, and one scale-scale. Partial, unlabelled, mismatched, and slope forms remain rejected. |
sd_phylo(species) ~ z |
Implemented for univariate Gaussian mu
|
This is the separate Box 1 direct-SD family: species-level predictors model the SD of a phylogenetic location random effect at observed tips. |
sd_phylo1(species) ~ z1 /
sd_phylo2(species) ~ z2
|
Implemented for bivariate mu1/mu2
|
Response-specific species-level predictors model the SD surfaces of
the two phylogenetic location random effects. This remains a Family B
direct-SD model, not a residual sigma1/sigma2
model and not a q=4 location-scale block. |
corpair(species, level = "phylogenetic", block = "p", from = "mu1", to = "mu2") ~ ecology |
Implemented for q=2 mu1/mu2
|
Species-level predictors model the same-species latent phylogenetic
location-location correlation through a positive-definite two-field
loading contract. corpairs(level = "phylogenetic") reports
the modelled response-scale mean, range, and number of species
values. |
spatial(1 | site, coords = coords) |
Implemented first slice | One trait has a coordinate-based spatial random intercept in the location predictor. This is a fixed coordinate covariance foundation, not the future mesh/SPDE path. |
spatial(1 + x | site, coords = coords) |
Implemented first one-slope slice | One trait has independent coordinate-spatial intercept and numeric slope fields in the location predictor. The two fields share the same coordinate precision, have separate SDs, and do not estimate an intercept-slope correlation. |
Matching spatial(1 | p | site, coords = coords) terms
in bivariate mu1 and mu2
|
Implemented first q=2 slice | Two Gaussian responses have correlated coordinate-spatial location
deviations. corpairs(level = "spatial") and
summary()$covariance report this mean-mean row separately
from residual rho12. |
Matching labelled spatial() terms in all four bivariate
mu1, mu2, sigma1, and
sigma2 formulas |
Implemented first q=4 slice | Two Gaussian responses have a constant coordinate-spatial location-scale covariance block with four endpoint SDs and six derived latent spatial correlations. |
phylo(1 | species, tree = tree) + spatial(1 | site, coords = coords)
in the same mu formula |
Planned | This is the intended third structural-dependence route, but the
fitter currently stops with a planned-feature message because multiple
structural mu layers need their own identifiability
checks. |
relmat(1 | id, K = K) or
relmat(1 | id, Q = Q)
|
Implemented first slice | A lower-level Gaussian mu random intercept for
user-supplied latent relatedness covariance or precision matrices. |
relmat(1 + x | id, K = K) or
relmat(1 + x | id, Q = Q)
|
Implemented first one-slope slice | One trait has independent known-matrix intercept and numeric slope fields in the location predictor. The two fields have separate SDs and do not estimate an intercept-slope correlation. |
Matching labelled relmat(1 | p | id, K = K) or
relmat(1 | p | id, Q = Q) terms in bivariate
mu1 and mu2
|
Implemented first q=2 slice | Two Gaussian responses have correlated lower-level known-relatedness
location deviations from the same matrix.
corpairs(level = "relmat") reports this mean-mean row
separately from residual rho12. |
spatial(1 | site, mesh = mesh) |
Planned | Mesh inputs will provide expert control for the scalable SPDE/GMRF spatial path. |
The fitted q=2 phylogenetic correlation-regression syntax is:
corpair(species, level = "phylogenetic", block = "p",
from = "mu1", to = "mu2") ~ ecologyThe predictor-dependent q=4 extensions remain planned:
# Planned q=4 extensions:
corpair(species, level = "phylogenetic", block = "p",
from = "mu1", to = "sigma1") ~ ecology
corpair(species, level = "phylogenetic", block = "p",
from = "mu1", to = "sigma2") ~ ecology
corpair(species, level = "phylogenetic", block = "p",
from = "mu2", to = "sigma1") ~ ecology
corpair(species, level = "phylogenetic", block = "p",
from = "mu2", to = "sigma2") ~ ecology
corpair(species, level = "phylogenetic", block = "p",
from = "sigma1", to = "sigma2") ~ ecologyThose formulae are not examples to run today. They mark the intended
next layer after constant phylogenetic corpairs() rows:
location-location, all four location-scale pairs, and scale-scale
correlation regression. The ordinary grouped corpair()
implementation cannot simply be copied here, because phylogenetic
effects share one tree-coupled covariance matrix rather than independent
per-species 2 by 2 blocks.
The first design contract for the location-location row uses two
independent unit phylogenetic fields. For species l, the
ecological predictor would define
rho_l = tanh(w_l^T alpha), then
c_l = sqrt((1 + rho_l) / 2) and
d_l = sqrt((1 - rho_l) / 2). The two location effects would
be
a1_l = tau1 (c_l z1_l + d_l z2_l)
a2_l = tau2 (c_l z1_l - d_l z2_l).This q=2 contract is for the location-location row only:
mu1-mu2. Location-scale rows such as
mu1-sigma2 and scale-scale rows such as
sigma1-sigma2 remain deferred until the q=4
version of this positive-definite construction is designed. This q=2
contract keeps the full covariance matrix positive definite and reduces
to the current constant bivariate phylogenetic covariance when all
rho_l are equal. When rho_l varies by species,
the model is nonstationary: it changes the tree-coupled latent
covariance surface, not just a residual correlation row.
A model ladder for two traits
The safest way to read the bivariate syntax is to build the model in layers. Each layer answers a different biological or statistical question.
1. Residual correlation only
Start here when the question is whether two measured traits remain correlated within the same observation after fixed effects:
fit_resid <- drmTMB(
drm_formula(
mu1 = heat_tolerance ~ climate,
mu2 = desiccation_tolerance ~ climate,
sigma1 = ~ habitat_variability,
sigma2 = ~ habitat_variability,
rho12 = ~ assay_context
),
family = c(gaussian(), gaussian()),
data = tolerance
)
rho12(fit_resid)This model has no phylogenetic random effects. The
rho12() output is the residual within-observation
correlation between heat tolerance and desiccation tolerance.
2. Add phylogenetic mean-mean correlation
Add matching phylo() terms to mu1 and
mu2 when the question is whether related species share
deviations in the two trait means:
fit_phylo_mean <- drmTMB(
drm_formula(
mu1 = heat_tolerance ~ climate + phylo(1 | p | species, tree = tree),
mu2 = desiccation_tolerance ~ climate +
phylo(1 | p | species, tree = tree),
sigma1 = ~ habitat_variability,
sigma2 = ~ habitat_variability,
rho12 = ~ assay_context
),
family = c(gaussian(), gaussian()),
data = tolerance
)
corpairs(fit_phylo_mean, level = "phylogenetic")
rho12(fit_phylo_mean)This model estimates one latent phylogenetic mean-mean correlation
and one residual rho12 surface. corpairs() and
rho12() answer different questions.
The coordinate-spatial sibling uses the same bivariate location layer but replaces the tree with a coordinate-derived precision:
fit_spatial_mean <- drmTMB(
drm_formula(
mu1 = heat_tolerance ~ climate +
spatial(1 | p | site, coords = coords),
mu2 = desiccation_tolerance ~ climate +
spatial(1 | p | site, coords = coords),
sigma1 = ~ habitat_variability,
sigma2 = ~ habitat_variability,
rho12 = ~ assay_context
),
family = c(gaussian(), gaussian()),
data = tolerance
)
corpairs(fit_spatial_mean, level = "spatial")
rho12(fit_spatial_mean)Use this model when nearby sites may share paired deviations in the
two response means. Use the q=4 spatial route only when the spatial
question also involves constant location-scale covariance across
mu1, mu2, sigma1, and
sigma2. For one-response models,
sigma ~ spatial(1 | site, coords = coords) fits an
intercept-only residual-scale spatial field, and
sigma ~ spatial(1 + x | site, coords = coords) has
point-fit/extractor evidence for the exact q1 one-slope route. Its
interval gate remains blocked. Predictor-dependent spatial
corpair() regression, multiple or labelled spatial sigma
slopes, and bivariate spatial slope blocks remain planned. Treat the
displayed spatial q2 correlation as a point estimate: interval
calibration for this intercept-only row is not yet validated.
3. Let species-level predictors change phylogenetic mean SDs
Use the Box 1 direct-SD family when the question is whether species-level predictors explain the magnitude of phylogenetic mean variation:
fit_phylo_sd <- drmTMB(
drm_formula(
mu1 = heat_tolerance ~ climate + phylo(1 | species, tree = tree),
mu2 = desiccation_tolerance ~ climate +
phylo(1 | species, tree = tree),
sigma1 = ~ habitat_variability,
sigma2 = ~ habitat_variability,
rho12 = ~ assay_context,
sd_phylo1(species) ~ habitat_variability_species,
sd_phylo2(species) ~ habitat_variability_species
),
family = c(gaussian(), gaussian()),
data = tolerance
)
coef(fit_phylo_sd, "sd_phylo1(species)")
coef(fit_phylo_sd, "sd_phylo2(species)")
corpairs(fit_phylo_sd, level = "phylogenetic")This is not a residual scale model. sd_phylo1() and
sd_phylo2() model the phylogenetic location SD surfaces for
the two responses. The cross-response phylogenetic mean-mean correlation
remains constant and is displayed as a point estimate; this direct-SD
route does not have validated interval evidence.
4. Move to a q=4 phylogenetic location-scale block
Use the q=4 block when the question is whether phylogenetic deviations in means and residual log-SDs move together:
fit_phylo_q4 <- drmTMB(
drm_formula(
mu1 = heat_tolerance ~ climate + phylo(1 | p | species, tree = tree),
mu2 = desiccation_tolerance ~ climate +
phylo(1 | p | species, tree = tree),
sigma1 = ~ habitat_variability +
phylo(1 | p | species, tree = tree),
sigma2 = ~ habitat_variability +
phylo(1 | p | species, tree = tree),
rho12 = ~ assay_context
),
family = c(gaussian(), gaussian()),
data = tolerance
)
corpairs(fit_phylo_q4, level = "phylogenetic")
rho12(fit_phylo_q4)This model estimates six latent phylogenetic correlations: one
location-location, four location-scale, and one scale-scale. These are
constant correlations in the phylogenetic random-effect covariance
block. The four location-scale rows are
mu1-sigma1,
mu1-sigma2,
mu2-sigma1, and
mu2-sigma2; the two cross-trait rows can be
biologically harder to explain, but they are statistically part of the
same q=4 block. They are not the residual rho12
surface.
The six q=4 correlations are point estimates. Direct or fix-and-refit profile intervals for these derived rows are not yet implemented and validated.
Read the six q=4 rows as latent phylogenetic covariance summaries:
| Pair | Biological reading | Common mistake |
|---|---|---|
mu1-mu2
|
species with positive phylogenetic deviations in trait 1 also tend to have positive or negative deviations in trait 2 | treating it as residual trait coupling |
mu1-sigma1
|
the evolutionary deviation in trait 1’s mean is associated with trait 1’s residual log-SD | reporting it as a fixed sigma1 slope |
mu1-sigma2
|
the evolutionary deviation in trait 1’s mean is associated with trait 2’s residual log-SD | dropping the cross-trait mean-scale row from the interpretation |
mu2-sigma1
|
the evolutionary deviation in trait 2’s mean is associated with trait 1’s residual log-SD | dropping the cross-trait mean-scale row from the interpretation |
mu2-sigma2
|
the evolutionary deviation in trait 2’s mean is associated with trait 2’s residual log-SD | treating it as the only mean-scale pair |
sigma1-sigma2
|
species-level residual-scale deviations co-vary across the two traits | treating it as residual rho12
|
The cross-trait rows are not optional bookkeeping. They are part of the fitted unstructured q=4 covariance block and should remain visible in examples even when the biological story is harder to tell.
5. Predictor-dependent latent phylogenetic correlations
The q=2 location-location route is fitted:
corpair(species, level = "phylogenetic", block = "p",
from = "mu1", to = "mu2") ~ ecologyThe q=4 endpoint-specific extensions are still planned:
# Planned q=4 extensions:
corpair(species, level = "phylogenetic", block = "p",
from = "mu1", to = "sigma1") ~ ecology
corpair(species, level = "phylogenetic", block = "p",
from = "mu1", to = "sigma2") ~ ecology
corpair(species, level = "phylogenetic", block = "p",
from = "mu2", to = "sigma1") ~ ecology
corpair(species, level = "phylogenetic", block = "p",
from = "mu2", to = "sigma2") ~ ecology
corpair(species, level = "phylogenetic", block = "p",
from = "sigma1", to = "sigma2") ~ ecologyThe fitted q=2 layer makes the phylogenetic location-location
random-effect correlation vary with a species-level ecological
predictor, analogous in spirit to rho12 = ~ assay_context
for residual correlation. The implementation is not identical to
residual rho12 regression: the phylogenetic likelihood must
still build one positive-definite covariance matrix for all species tied
together by the tree. The selected q=2 design uses a two-field loading
construction, so the modelled rho_l is the same-species
phylogenetic correlation while between-species covariances remain tied
to the tree and become nonstationary when rho_l varies.
This first implementation target is only the location-location row;
location-scale and scale-scale correlation regressions are q=4
extensions and remain deferred. The explicit
level = "phylogenetic" keeps this layer separate from
ordinary species effects and future spatial effects. Endpoint-specific
from / to syntax avoids treating all four
location-scale pairs as if they had the same biological meaning. Use
corpairs() for fitted constant or modelled latent
phylogenetic correlations and rho12() for fitted residual
correlations. The ordinary group-level q=2 analogue is:
corpair(id, level = "group", block = "p", from = "mu1", to = "mu2") ~ w
models the correlation between two labelled ordinary location random
intercepts.
What should get 95% intervals?
There are two different kinds of “random-effect” uncertainty.
The first kind is uncertainty in the covariance parameters:
random-effect SDs and correlations. These are the quantities shown by
sdpars, corpars, corpairs(), and
summary(fit)$covariance. The admitted interval method is
row-specific rather than a blanket preference for profile likelihood.
The q1 mu anchors and the exact phylo/relmat slope-only q2
mu1:x/mu2:x SD rows use the default
location-axis bias-corrected, small-sample-t Wald channel; q1
sigma uses raw uncorrected log-SD Wald-z evidence and
treats profile as diagnostic-only at g = 8; the Arc 1a REML
cells use direct structured-SD profiles only in their tested discrete
domains. Check the capability-and-limits
guide for the evidence channel attached to a particular row.
The intercept-only q2 example on this page is not one of those admitted slope-only rows. Its default Wald output is diagnostic rather than a validated reporting interval:
confint(fit_phylo_mean, parm = "variance_components")The direct Wald route uses the fitted log-SD scale for SD targets and
the guarded atanh correlation-link scale for direct correlation targets
before returning intervals on the reported scale. For this
intercept-only q2 row, neither a finite Wald result nor an available
target promotes the row beyond diagnostic-only. A targeted profile can
diagnose boundary geometry when profile_targets(fit) lists
the target, but it does not replace a validated channel or promote a row
whose interval evidence is diagnostic-only.
For a diagnostic look at likelihood geometry, use
profile_targets() to find the target name and
confint(..., method = "profile") to request a diagnostic
profile. The intercept-only bivariate q2 example below is
diagnostic-only under both Wald and profile channels:
profile_targets(fit_phylo_mean, ready_only = TRUE)
phylo_cor <- "cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | phylo | species)"
confint(
fit_phylo_mean,
parm = phylo_cor,
method = "profile",
profile_precision = "fast"
)Diagnostic profile rows include conf.status = "profile".
They also include profile.boundary and
profile.message, so an interval that reaches a lower SD
boundary or approaches a correlation boundary is marked. Do not report
either interval above as validated for this intercept-only q2 fit. The
admitted q2 reporting channel belongs only to the exact phylo/relmat
slope-only SD rows named in the capability guide.
profile_precision = "fast" is a quicker diagnostic pass
that uses coarser TMB::tmbprofile() controls; use default
precision only when a more detailed geometry diagnosis is needed.
The second kind is uncertainty in conditional random-effect modes, such as the species-specific fitted deviations. Those intervals are useful for caterpillar plots and diagnostics, but they are not the same as intervals for phylogenetic SDs or correlations. The current priority is covariance-parameter intervals first, then conditional-mode uncertainty once the covariance interfaces are stable.
For q=4 ordinary or phylogenetic blocks,
profile_targets() lists the six correlations as derived
unstructured-correlation targets. They are reported as point estimates
today, but direct profile intervals for all six rows are not claimed
yet. The first ordinary and phylogenetic corpair() ~ w
implementations are narrower: they fit only the q=2
mu1-mu2 location-location pair for matching
labelled ordinary or phylogenetic random intercepts. Those direct
correlation-regression paths expose link-scale fixed-effect coefficients
through coef(), summary(),
vcov(), and profile_targets(), while
corpairs() reports the response-scale mean, minimum,
maximum, and number of group-level or species-level correlation values.
The following call is a geometry diagnostic at a chosen group-level
predictor value; profile availability is not validated interval evidence
for this row:
confint(
fit_ord_corpair,
parm = 'corpair(id, level = "group", block = "p", from = "mu1", to = "mu2")',
method = "profile",
newdata = data.frame(w = 0)
)The animal q=2 path uses the same fitted-versus-planned distinction.
Constant corpairs(level = "animal") rows are fitted for
matching labelled
animal(1 | p | individual, pedigree = pedigree),
A, or Ainv terms, but predictor-dependent
animal corpair() regression and sparse large-pedigree
precision construction remain planned.
The phylogenetic q=2 path exposes the same diagnostic profiling
interface, but the parm names the phylogenetic level:
confint(
fit_phylo_corpair,
parm = paste0(
'corpair(species, level = "phylogenetic", block = "p", ',
'from = "mu1", to = "mu2")'
),
method = "profile",
newdata = data.frame(ecology = 0)
)corpairs(conf.int = TRUE) still reports
newdata_required for the modelled summary row because that
row averages and ranges over group-level or species-level correlation
values. Neither predictor-dependent q2 example above has coverage-backed
interval validation, so do not use those diagnostic profiles as
reporting intervals. q=4 rows still need a fix-and-refit or
reparameterized profile before 95% intervals are claimed for all six
derived correlations.
Mathematical contract
The general structured-effect bridge is:
Here
is the distributional parameter being modelled. For the simplest fitted
examples,
:
the structured effect is added to the mean predictor. Current Gaussian
first slices also include intercept-only structured sigma
terms, q1 sigma one-slope routes for all four providers,
and selected bivariate covariance blocks. The phylo, A-matrix animal,
and K/Q relmat sigma slopes are inference-ready with caveats; the
spatial sigma-slope interval gate, additional multiple or labelled
structured-sigma layouts outside the exact fitted ledger cells, and
structured rho12 remain planned. For phylogenetic models,
is a tree-derived phylogenetic covariance matrix
.
For spatial models,
will be a distance-derived matrix or an SPDE/GMRF precision.
For the implemented univariate Gaussian phylogenetic location model:
The phylogenetic SD
measures how strongly related species remain similar in their fitted
mean after the fixed effects have been included. The residual
sigma remains the within-observation residual SD. These are
different scale parameters.
The matching implemented R syntax is:
drmTMB(
drm_formula(
y ~ x1 + phylo(1 | species, tree = tree),
sigma ~ x1
),
family = gaussian(),
data = dat
)Worked example: thermal tolerance across species
Suppose we measure thermal tolerance for multiple observations per
species and ask whether body size predicts thermal tolerance after
accounting for shared ancestry. In a real analysis, tree
would usually come from a phylogenetic data workflow. For this vignette,
the setup code creates a small ultrametric branch-length tree so the
example stays self-contained.
set.seed(31)
tree <- balanced_ultrametric_tree(16)
species_info <- data.frame(
species = tree$tip.label,
body_size = scale(
rnorm(16, mean = rep(c(-0.4, 0.4), each = 8), sd = 0.35)
)[, 1]
)
phylo_shift <- rep(c(-0.5, 0.5), each = 8) +
rep(c(-0.18, 0.18), each = 4, length.out = 16)
n_each <- 5
dat <- species_info[rep(seq_len(nrow(species_info)), each = n_each), ]
dat$body_size <- dat$body_size + rnorm(nrow(dat), sd = 0.08)
dat$thermal_tolerance <- 1.2 + 0.45 * dat$body_size +
phylo_shift[match(dat$species, species_info$species)] +
rnorm(nrow(dat), sd = 0.25)
head(dat)
#> species body_size thermal_tolerance
#> 1 sp_1 -1.296481 -0.09668096
#> 1.1 sp_1 -1.224245 0.37697808
#> 1.2 sp_1 -1.290090 0.24284232
#> 1.3 sp_1 -1.366560 -0.06074780
#> 1.4 sp_1 -1.424426 -0.09886188
#> 2 sp_2 -1.618790 -0.07958876The fitted model follows the equation above:
fit_phylo <- drmTMB(
drm_formula(
thermal_tolerance ~ body_size + phylo(1 | species, tree = tree),
sigma ~ 1
),
family = gaussian(),
data = dat
)
summary(fit_phylo)
#> <summary.drmTMB>
#> estimator: ML
#> estimate std_error
#> mu:(Intercept) 1.2055884 0.14702564
#> mu:body_size 0.4899816 0.06517078
#> sigma:(Intercept) -1.4293873 0.08571324
#> Distributional, random-effect, scale, and correlation parameters:
#> component dpar term
#> sigma distributional-scale sigma (constant)
#> sd:mu:phylo(1 | species) random-effect-sd mu phylo(1 | species)
#> estimate std_error scale
#> sigma 0.2394556 0.02052451 response
#> sd:mu:phylo(1 | species) 0.2986174 0.05841679 response
#> Derived summaries:
#> quantity level group
#> derived:phylogenetic_signal(species) phylogenetic_signal phylogenetic species
#> term estimate
#> derived:phylogenetic_signal(species) phylo(1 | species) 0.6086378
#> random_effect_variance residual_variance
#> derived:phylogenetic_signal(species) 0.08917232 0.05733898
#> logLik: -13.41
#> convergence: 0How to read this output:
The mu:body_size row is the fixed-effect slope for body
size in the mean thermal tolerance model. The
sigma:(Intercept) row is the log residual SD, so
exp(coef(fit_phylo, "sigma")) gives the within-observation
residual SD. The phylo(1 | species) random-effect SD is
,
the phylogenetic among-species SD in the mean after body size has been
included.
phylo_sd <- unname(fit_phylo$sdpars$mu["phylo(1 | species)"])
residual_sd <- exp(coef(fit_phylo, "sigma")["(Intercept)"])
c(phylogenetic_sd = phylo_sd, residual_sd = residual_sd)
#> phylogenetic_sd residual_sd.(Intercept)
#> 0.2986174 0.2394556In this run, related species remain similar in thermal tolerance after accounting for body size. The fitted phylogenetic SD is larger than the within-observation residual SD, so shared ancestry explains a visible part of the remaining mean-level structure.
check_drm() reports whether the optimizer, Hessian,
scale, dropped-row, and phylogenetic replication checks passed:
check_drm(fit_phylo)
#> <drm_check: 15 checks>
#> ok: 15; notes: 0; warnings: 0; errors: 0
#> check status
#> optimizer_convergence ok
#> optimizer_budget ok
#> finite_objective ok
#> logsigma_clamp_active ok
#> fixed_gradient ok
#> sdreport_status ok
#> hessian_positive_definite ok
#> standard_errors_finite ok
#> standard_errors_inflated ok
#> dropped_rows ok
#> positive_scale ok
#> random_effect_sd_boundary ok
#> fixed_effect_design_size ok
#> phylo_mu_replication ok
#> phylo_mu_diagnostics ok
#> value
#> 0
#> iterations=19; function=28; gradient=20
#> 13.41
#> <NA>
#> max=0.0000003639; component=beta_mu[2]
#> ok
#> TRUE
#> range=[0.06517,0.1470]
#> n_inflated=0; max_se=0.1470; median_se=0.08571
#> nobs=80; dropped=0
#> min=0.2395
#> min=0.2986; boundary=0.0001000; term=mu.phylo(1 | species)
#> total_mb=0.01305; max_cols=2; largest=mu; largest_class=matrix; largest_density=1.000
#> min_species_n=5
#> group=species; n_species=16; min_species_n=5; phylo_sd=0.2986; sd_ratio=1.247
#> message
#> nlminb convergence code is 0.
#> Optimizer evaluation counts recorded; no eval.max or iter.max control was supplied.
#> Objective and log-likelihood are finite.
#> The log(sigma) clamp is not active at the optimum.
#> Maximum absolute fixed gradient is <= 0.001; largest component is beta_mu[2].
#> TMB::sdreport() completed successfully.
#> sdreport reports a positive-definite Hessian.
#> All fixed-effect standard errors are finite.
#> No fixed-effect standard error is inflated relative to the others.
#> No rows were dropped by model-frame or known-covariance filtering.
#> All fitted scale values are finite and positive.
#> All fitted random-effect standard deviations are finite, positive, and above the requested lower-boundary warning threshold.
#> Dense fixed-effect design matrices are modest for this fit.
#> Every observed species has at least two fitted observations.
#> The phylogenetic random effect has replicated species and a non-negligible fitted SD relative to residual scale.Practical checks for phylogenetic fits
The public phylo() term requires an ultrametric tree
with branch lengths. Users pass the tree; drmTMB builds the
sparse phylogenetic precision internally. You do not need to construct
the phylogenetic covariance matrix or A-inverse yourself.
If a phylogenetic fit fails before optimization, check these points first:
- the tree object has class
phylo; - every observed species in
data$speciesappears intree$tip.label; - species names use the same spelling and underscore/space convention in the data and tree;
- the tree has finite positive branch lengths and is ultrametric;
- the model uses an implemented syntax: Gaussian
phylo()terms in the documented univariate or matching bivariate formulas, ordinary Poisson/NB2 q1 phylogeneticmuintercept-plus-one-slope terms, recovery-grade NB2 q1 phylogeneticsigma, Student-t q1 phylogeneticnu, or cumulative-logit q1 phylogeneticmugates recorded in the live ledger.
Additional multiple or labelled phylogenetic-slope layouts outside
the exact fitted ledger cells, phylogenetic slope correlations,
non-Gaussian phylogenetic effects outside those exact gates, and
structured effects in rho12 remain planned. Partial,
unlabelled, mismatched, and slope q=4 phylogenetic location-scale forms
should error clearly rather than silently fitting a different model.
Bivariate phylogenetic correlations: model layer by layer
For two Gaussian responses, drmTMB separates the
residual correlation from the phylogenetic random-effect correlation.
Let
.
A bivariate phylogenetic location model can be written as:
The residual coscale formula, rho12 = ~ assay_context,
models
in
.
This is an observation-level residual correlation.
The phylogenetic location effects are a separate latent layer. Let and collect the species-level phylogenetic location effects for responses 1 and 2. If is the tree-derived species correlation matrix, then:
Here
is the phylogenetic mean-mean correlation. It is reported by
corpairs(fit, level = "phylogenetic"), not by
rho12().
The matching R syntax is:
fit_biv_phylo <- drmTMB(
drm_formula(
mu1 = heat_tolerance ~ climate + phylo(1 | p | species, tree = tree),
mu2 = desiccation_tolerance ~ climate +
phylo(1 | p | species, tree = tree),
sigma1 = ~ habitat_variability,
sigma2 = ~ habitat_variability,
rho12 = ~ assay_context
),
family = c(gaussian(), gaussian()),
data = tolerance
)
corpairs(fit_biv_phylo, level = "phylogenetic")
rho12(fit_biv_phylo)In that output, corpairs() gives the constant
phylogenetic correlation point estimate among species-level mean
deviations. rho12() gives the fitted residual correlation
for each observation, which can vary with assay_context.
The intercept-only q2 row does not have validated interval evidence; a
callable profile is a diagnostic, not an inference-ready claim.
This is the key implementation boundary. drmTMB can
estimate latent phylogenetic correlations when the matching
phylo() terms define a shared covariance block. It can also
fit the first predictor-dependent latent phylogenetic correlation
formula for the q=2 location-location row. In other words,
rho12 = ~ assay_context is implemented for residual
correlation regression, and
corpair(species, level = "phylogenetic", block = "p", from = "mu1", to = "mu2") ~ ecology
is implemented for species-level predictors beside matching labelled
mu1 and mu2 phylo() terms. The
q=4 location-scale and scale-scale correlation regressions remain
planned.
The q=4 phylogenetic location-scale block extends the same idea to four endpoints. For endpoint , let be the phylogenetic effect for species . The covariance contract is:
The four linear predictors then contain one endpoint-specific phylogenetic effect:
The fitted
is written as
,
where
contains four phylogenetic SDs and
contains six latent correlations. Those six correlations are the
corpairs(fit, level = "phylogenetic") rows: one
location-location row, four location-scale rows, and one scale-scale
row. The four location-scale rows are
-,
-,
-,
and
-.
Worked example: two traits with phylogenetic location-scale covariance
Now suppose the scientific question is about two tolerance traits measured repeatedly for the same species. We want climate to explain each trait mean, habitat variability to explain each residual SD, and assay context to explain the residual within-observation correlation. We also want the species-level phylogenetic deviations in both means and both log-SDs to share one latent q=4 covariance block.
The simulated data below are deliberately small enough for the
article to run. The hidden setup creates the tree and species-level
phylogenetic effects; the fitted model uses only public
drmTMB() syntax.
sim_q4 <- simulate_phylo_q4_traits()
tree <- sim_q4$tree
tol_dat <- sim_q4$data
head(tol_dat)
#> species climate habitat_variability assay_context heat_tolerance
#> 1 sp_1 0.8536583 1.3302474 -0.5 -1.26256225
#> 2 sp_1 1.4302245 0.7398460 -0.5 -0.25651175
#> 3 sp_1 2.2607858 0.9024009 -0.5 1.49317129
#> 4 sp_1 1.1587544 -1.0788332 0.5 -0.13889853
#> 5 sp_1 0.4185905 -0.6995513 0.5 0.24863253
#> 6 sp_1 -0.2717992 0.1264835 0.5 -0.07806504
#> desiccation_tolerance
#> 1 -1.01377546
#> 2 0.05515596
#> 3 -0.71601270
#> 4 -0.55595513
#> 5 -0.22743664
#> 6 -0.04126080The model uses the same labelled phylo() term in all
four endpoints:
fit_phylo_q4 <- suppressWarnings(
drmTMB(
drm_formula(
mu1 = heat_tolerance ~ climate + phylo(1 | p | species, tree = tree),
mu2 = desiccation_tolerance ~ climate +
phylo(1 | p | species, tree = tree),
sigma1 = ~ habitat_variability +
phylo(1 | p | species, tree = tree),
sigma2 = ~ habitat_variability +
phylo(1 | p | species, tree = tree),
rho12 = ~ assay_context
),
family = c(gaussian(), gaussian()),
data = tol_dat,
control = list(eval.max = 1000, iter.max = 1000)
)
)
summary(fit_phylo_q4)
#> <summary.drmTMB>
#> estimator: ML
#> estimate std_error std_error.status
#> mu1:(Intercept) 0.12404011 NA sdreport_non_pd_hessian
#> mu1:climate 0.25588984 NA sdreport_non_pd_hessian
#> mu2:(Intercept) -0.36344078 NA sdreport_non_pd_hessian
#> mu2:climate -0.25668869 NA sdreport_non_pd_hessian
#> sigma1:(Intercept) -1.04050450 NA sdreport_non_pd_hessian
#> sigma1:habitat_variability 0.13799113 NA sdreport_non_pd_hessian
#> sigma2:(Intercept) -1.31598087 NA sdreport_non_pd_hessian
#> sigma2:habitat_variability -0.12081564 NA sdreport_non_pd_hessian
#> rho12:(Intercept) 0.09221661 NA sdreport_non_pd_hessian
#> rho12:assay_context -0.15289535 NA sdreport_non_pd_hessian
#> Distributional, random-effect, scale, and correlation parameters:
#> component
#> sd:mu:mu1:phylo(1 | p | species) random-effect-sd
#> sd:mu:mu2:phylo(1 | p | species) random-effect-sd
#> sd:mu:sigma1:phylo(1 | p | species) random-effect-sd
#> sd:mu:sigma2:phylo(1 | p | species) random-effect-sd
#> cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | p | species) random-effect-correlation
#> cor:phylo:cor(mu1:(Intercept),sigma1:(Intercept) | p | species) random-effect-correlation
#> cor:phylo:cor(mu1:(Intercept),sigma2:(Intercept) | p | species) random-effect-correlation
#> cor:phylo:cor(mu2:(Intercept),sigma1:(Intercept) | p | species) random-effect-correlation
#> cor:phylo:cor(mu2:(Intercept),sigma2:(Intercept) | p | species) random-effect-correlation
#> cor:phylo:cor(sigma1:(Intercept),sigma2:(Intercept) | p | species) random-effect-correlation
#> fitted:sigma1 distributional-scale
#> fitted:sigma2 distributional-scale
#> fitted:rho12 residual-correlation
#> dpar
#> sd:mu:mu1:phylo(1 | p | species) mu
#> sd:mu:mu2:phylo(1 | p | species) mu
#> sd:mu:sigma1:phylo(1 | p | species) mu
#> sd:mu:sigma2:phylo(1 | p | species) mu
#> cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | p | species) phylo
#> cor:phylo:cor(mu1:(Intercept),sigma1:(Intercept) | p | species) phylo
#> cor:phylo:cor(mu1:(Intercept),sigma2:(Intercept) | p | species) phylo
#> cor:phylo:cor(mu2:(Intercept),sigma1:(Intercept) | p | species) phylo
#> cor:phylo:cor(mu2:(Intercept),sigma2:(Intercept) | p | species) phylo
#> cor:phylo:cor(sigma1:(Intercept),sigma2:(Intercept) | p | species) phylo
#> fitted:sigma1 sigma1
#> fitted:sigma2 sigma2
#> fitted:rho12 rho12
#> term
#> sd:mu:mu1:phylo(1 | p | species) mu1:phylo(1 | p | species)
#> sd:mu:mu2:phylo(1 | p | species) mu2:phylo(1 | p | species)
#> sd:mu:sigma1:phylo(1 | p | species) sigma1:phylo(1 | p | species)
#> sd:mu:sigma2:phylo(1 | p | species) sigma2:phylo(1 | p | species)
#> cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | p | species) cor(mu1:(Intercept),mu2:(Intercept) | p | species)
#> cor:phylo:cor(mu1:(Intercept),sigma1:(Intercept) | p | species) cor(mu1:(Intercept),sigma1:(Intercept) | p | species)
#> cor:phylo:cor(mu1:(Intercept),sigma2:(Intercept) | p | species) cor(mu1:(Intercept),sigma2:(Intercept) | p | species)
#> cor:phylo:cor(mu2:(Intercept),sigma1:(Intercept) | p | species) cor(mu2:(Intercept),sigma1:(Intercept) | p | species)
#> cor:phylo:cor(mu2:(Intercept),sigma2:(Intercept) | p | species) cor(mu2:(Intercept),sigma2:(Intercept) | p | species)
#> cor:phylo:cor(sigma1:(Intercept),sigma2:(Intercept) | p | species) cor(sigma1:(Intercept),sigma2:(Intercept) | p | species)
#> fitted:sigma1 fitted range
#> fitted:sigma2 fitted range
#> fitted:rho12 fitted range
#> estimate
#> sd:mu:mu1:phylo(1 | p | species) 0.56283742
#> sd:mu:mu2:phylo(1 | p | species) 0.46546831
#> sd:mu:sigma1:phylo(1 | p | species) 0.29622384
#> sd:mu:sigma2:phylo(1 | p | species) 0.17803706
#> cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | p | species) 0.57228119
#> cor:phylo:cor(mu1:(Intercept),sigma1:(Intercept) | p | species) -0.08609142
#> cor:phylo:cor(mu1:(Intercept),sigma2:(Intercept) | p | species) -0.31735908
#> cor:phylo:cor(mu2:(Intercept),sigma1:(Intercept) | p | species) -0.13720281
#> cor:phylo:cor(mu2:(Intercept),sigma2:(Intercept) | p | species) 0.48759390
#> cor:phylo:cor(sigma1:(Intercept),sigma2:(Intercept) | p | species) 0.41827625
#> fitted:sigma1 0.33801985
#> fitted:sigma2 0.26609205
#> fitted:rho12 0.09142517
#> minimum
#> sd:mu:mu1:phylo(1 | p | species) NA
#> sd:mu:mu2:phylo(1 | p | species) NA
#> sd:mu:sigma1:phylo(1 | p | species) NA
#> sd:mu:sigma2:phylo(1 | p | species) NA
#> cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | p | species) NA
#> cor:phylo:cor(mu1:(Intercept),sigma1:(Intercept) | p | species) NA
#> cor:phylo:cor(mu1:(Intercept),sigma2:(Intercept) | p | species) NA
#> cor:phylo:cor(mu2:(Intercept),sigma1:(Intercept) | p | species) NA
#> cor:phylo:cor(mu2:(Intercept),sigma2:(Intercept) | p | species) NA
#> cor:phylo:cor(sigma1:(Intercept),sigma2:(Intercept) | p | species) NA
#> fitted:sigma1 0.14051402
#> fitted:sigma2 0.15222674
#> fitted:rho12 0.01576761
#> maximum
#> sd:mu:mu1:phylo(1 | p | species) NA
#> sd:mu:mu2:phylo(1 | p | species) NA
#> sd:mu:sigma1:phylo(1 | p | species) NA
#> sd:mu:sigma2:phylo(1 | p | species) NA
#> cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | p | species) NA
#> cor:phylo:cor(mu1:(Intercept),sigma1:(Intercept) | p | species) NA
#> cor:phylo:cor(mu1:(Intercept),sigma2:(Intercept) | p | species) NA
#> cor:phylo:cor(mu2:(Intercept),sigma1:(Intercept) | p | species) NA
#> cor:phylo:cor(mu2:(Intercept),sigma2:(Intercept) | p | species) NA
#> cor:phylo:cor(sigma1:(Intercept),sigma2:(Intercept) | p | species) NA
#> fitted:sigma1 0.6191785
#> fitted:sigma2 0.4034128
#> fitted:rho12 0.1670827
#> scale
#> sd:mu:mu1:phylo(1 | p | species) response
#> sd:mu:mu2:phylo(1 | p | species) response
#> sd:mu:sigma1:phylo(1 | p | species) response
#> sd:mu:sigma2:phylo(1 | p | species) response
#> cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | p | species) response
#> cor:phylo:cor(mu1:(Intercept),sigma1:(Intercept) | p | species) response
#> cor:phylo:cor(mu1:(Intercept),sigma2:(Intercept) | p | species) response
#> cor:phylo:cor(mu2:(Intercept),sigma1:(Intercept) | p | species) response
#> cor:phylo:cor(mu2:(Intercept),sigma2:(Intercept) | p | species) response
#> cor:phylo:cor(sigma1:(Intercept),sigma2:(Intercept) | p | species) response
#> fitted:sigma1 response
#> fitted:sigma2 response
#> fitted:rho12 response
#> Random-effect covariance summaries:
#> level group block from_dpar to_dpar class correlation
#> 1 phylogenetic species p mu1 mu2 mean-mean 0.57228119
#> 2 phylogenetic species p mu1 sigma1 mean-scale -0.08609142
#> 3 phylogenetic species p mu1 sigma2 mean-scale -0.31735908
#> 4 phylogenetic species p mu2 sigma1 mean-scale -0.13720281
#> 5 phylogenetic species p mu2 sigma2 mean-scale 0.48759390
#> 6 phylogenetic species p sigma1 sigma2 scale-scale 0.41827625
#> from_sd to_sd covariance
#> 1 0.5628374 0.4654683 0.14992793
#> 2 0.5628374 0.2962238 -0.01435367
#> 3 0.5628374 0.1780371 -0.03180126
#> 4 0.4654683 0.2962238 -0.01891791
#> 5 0.4654683 0.1780371 0.04040720
#> 6 0.2962238 0.1780371 0.02205940
#> logLik: -173.1
#> convergence: 1The fixed-effect rows answer the distributional-regression questions.
The mu1:climate and mu2:climate rows are mean
effects. The sigma1:habitat_variability and
sigma2:habitat_variability rows are effects on log residual
SD. The rho12:assay_context row is the residual coscale
effect: it changes the residual correlation between the two traits
within the same observation.
The phylogenetic q=4 block is a different layer. Use
corpairs() to read the latent species-level
correlations:
corpairs(fit_phylo_q4, level = "phylogenetic")
#> level group block from_dpar to_dpar from_coef to_coef
#> 1 phylogenetic species p mu1 mu2 (Intercept) (Intercept)
#> 2 phylogenetic species p mu1 sigma1 (Intercept) (Intercept)
#> 3 phylogenetic species p mu1 sigma2 (Intercept) (Intercept)
#> 4 phylogenetic species p mu2 sigma1 (Intercept) (Intercept)
#> 5 phylogenetic species p mu2 sigma2 (Intercept) (Intercept)
#> 6 phylogenetic species p sigma1 sigma2 (Intercept) (Intercept)
#> from_response to_response class
#> 1 heat_tolerance desiccation_tolerance mean-mean
#> 2 heat_tolerance heat_tolerance mean-scale
#> 3 heat_tolerance desiccation_tolerance mean-scale
#> 4 desiccation_tolerance heat_tolerance mean-scale
#> 5 desiccation_tolerance desiccation_tolerance mean-scale
#> 6 heat_tolerance desiccation_tolerance scale-scale
#> parameter estimate
#> 1 cor(mu1:(Intercept),mu2:(Intercept) | p | species) 0.57228119
#> 2 cor(mu1:(Intercept),sigma1:(Intercept) | p | species) -0.08609142
#> 3 cor(mu1:(Intercept),sigma2:(Intercept) | p | species) -0.31735908
#> 4 cor(mu2:(Intercept),sigma1:(Intercept) | p | species) -0.13720281
#> 5 cor(mu2:(Intercept),sigma2:(Intercept) | p | species) 0.48759390
#> 6 cor(sigma1:(Intercept),sigma2:(Intercept) | p | species) 0.41827625
#> min max n_values link_estimate link_min link_max
#> 1 0.57228119 0.57228119 1 0.65090926 0.65090926 0.65090926
#> 2 -0.08609142 -0.08609142 1 -0.08630515 -0.08630515 -0.08630515
#> 3 -0.31735908 -0.31735908 1 -0.32870802 -0.32870802 -0.32870802
#> 4 -0.13720281 -0.13720281 1 -0.13807373 -0.13807373 -0.13807373
#> 5 0.48759390 0.48759390 1 0.53289954 0.53289954 0.53289954
#> 6 0.41827625 0.41827625 1 0.44560142 0.44560142 0.44560142
#> modelled conf.status interval_source
#> 1 FALSE not_requested not_available
#> 2 FALSE not_requested not_available
#> 3 FALSE not_requested not_available
#> 4 FALSE not_requested not_available
#> 5 FALSE not_requested not_available
#> 6 FALSE not_requested not_availableThose six rows are not residual correlations. The
mean-mean row asks whether species that are
phylogenetically high for heat tolerance also tend to be high for
desiccation tolerance. The four mean-scale rows ask whether
evolutionary deviations in means are associated with evolutionary
deviations in variability: same-trait pairs
(mu1-sigma1,
mu2-sigma2) and cross-trait pairs
(mu1-sigma2,
mu2-sigma1). The cross-trait rows are
statistically valid q=4 covariance rows, but their biological
interpretation should be written with extra care.
The scale-scale row asks whether the two traits’
phylogenetic log-SD deviations move together. These q=4 rows are point
estimates and remain derived targets until a direct or fix-and-refit
profile is implemented and validated.
For a compact scalar view of the residual layer, use
rho12():
check_drm() keeps these layers separate. In small
examples, q=4 models can warn about optimizer convergence even when
gradients, Hessian status, and q=4 covariance diagnostics are
acceptable. Treat this article fit as an output map; for scientific
inference, inspect the full diagnostic table and use larger datasets or
simpler covariance structure when needed.
q4_check <- check_drm(fit_phylo_q4)
q4_check[q4_check$check %in% c(
"optimizer_convergence",
"fixed_gradient",
"hessian_positive_definite",
"biv_phylo_q4_covariance"
), ]
#> <drm_check: 4 checks>
#> ok: 2; notes: 0; warnings: 2; errors: 0
#> check status
#> optimizer_convergence warning
#> fixed_gradient ok
#> hessian_positive_definite warning
#> biv_phylo_q4_covariance ok
#> value
#> 1
#> max=0.00007279; component=beta_mu2[2]
#> FALSE
#> group=species; block=p; covariance_mode=unstructured; q=4; n_species=32; min_species_n=6; min_location_sd_ratio=1.665; min_log_sigma_sd=0.1780; max_abs_cor=0.5723; boundary=0.9800
#> message
#> nlminb convergence message: singular convergence (7)
#> Maximum absolute fixed gradient is <= 0.001; largest component is beta_mu2[2].
#> sdreport does not report a positive-definite Hessian.
#> Phylogenetic q4 location-scale covariance has replicated levels, non-negligible fitted component SDs, and latent correlations away from the boundary.The fitted q=4 correlations are point summaries. They are listed in
profile_targets() as derived theta_phylo
targets, not as direct profile-ready intervals:
subset(
profile_targets(fit_phylo_q4),
startsWith(parm, "cor:phylo:"),
select = c(parm, target_type, profile_ready, profile_note)
)
#> parm
#> 15 cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | p | species)
#> 16 cor:phylo:cor(mu1:(Intercept),sigma1:(Intercept) | p | species)
#> 17 cor:phylo:cor(mu1:(Intercept),sigma2:(Intercept) | p | species)
#> 18 cor:phylo:cor(mu2:(Intercept),sigma1:(Intercept) | p | species)
#> 19 cor:phylo:cor(mu2:(Intercept),sigma2:(Intercept) | p | species)
#> 20 cor:phylo:cor(sigma1:(Intercept),sigma2:(Intercept) | p | species)
#> target_type profile_ready profile_note
#> 15 derived FALSE derived_unstructured_correlation
#> 16 derived FALSE derived_unstructured_correlation
#> 17 derived FALSE derived_unstructured_correlation
#> 18 derived FALSE derived_unstructured_correlation
#> 19 derived FALSE derived_unstructured_correlation
#> 20 derived FALSE derived_unstructured_correlationThe q=2 predictor-dependent latent phylogenetic correlation is fitted
for the location-location row. The extractor is corpairs(),
and the formula syntax is singular:
corpair(species, level = "phylogenetic", block = "p",
from = "mu1", to = "mu2") ~ wThe predictor-dependent q=4 extensions remain planned:
# Planned q=4 extensions:
corpair(species, level = "phylogenetic", block = "p",
from = "mu1", to = "sigma1") ~ w
corpair(species, level = "phylogenetic", block = "p",
from = "mu1", to = "sigma2") ~ w
corpair(species, level = "phylogenetic", block = "p",
from = "mu2", to = "sigma1") ~ w
corpair(species, level = "phylogenetic", block = "p",
from = "mu2", to = "sigma2") ~ w
corpair(species, level = "phylogenetic", block = "p",
from = "sigma1", to = "sigma2") ~ wThis endpoint naming matters because the cross-trait mean-scale pairs
can be biologically harder to interpret than same-trait pairs. Use
rho12 = ~ assay_context for residual correlation
predictors. Use corpairs(fit, level = "phylogenetic") to
extract fitted constant or modelled latent phylogenetic
correlations.
Implemented third route: coordinate spatial intercepts and one slope
Spatial fields are the third route in the structural-dependence
ladder. They are deliberately shaped to look parallel to phylogenetic
effects, but they use site coordinates rather than a tree. The first
fitted paths are coordinate-based univariate Gaussian mu
coefficient fields: a random intercept, and one independent random slope
for a numeric predictor. The example below keeps the data deliberately
small so the models can be rebuilt inside the article:
spatial_example <- simulate_spatial_location_data()
spatial_dat <- spatial_example$data
coords <- spatial_example$coords
fit_spatial <- drmTMB(
drm_formula(
y ~ depth + temp + spatial(1 | site, coords = coords),
sigma ~ depth
),
family = gaussian(),
data = spatial_dat
)The fitted spatial SD is a structured location-scale component, not
the residual sigma. The conditional spatial effects are
site-level deviations in the location predictor, after the fixed effects
and residual scale have been estimated:
fit_spatial$sdpars$mu
#> spatial(1 | site)
#> 0.3808036
head(ranef(fit_spatial, "spatial_mu")$terms[[1]])
#> site_1 site_2 site_3 site_4 site_5 site_6
#> -0.2640539 -0.4642452 -0.0359260 -0.1560390 -0.3023818 0.2240647
profile_targets(fit_spatial, ready_only = TRUE)[
grep("spatial", profile_targets(fit_spatial, ready_only = TRUE)$parm),
c("parm", "estimate", "profile_ready")
]
#> parm estimate profile_ready
#> 6 sd:mu:spatial(1 | site) 0.3808036 TRUEWhen the biological question is whether a local environmental
gradient has a different slope in different parts of the sampled
landscape, add one numeric coefficient inside the spatial()
term:
fit_spatial_slope <- drmTMB(
drm_formula(
y ~ depth + temp + spatial(1 + depth | site, coords = coords),
sigma ~ depth
),
family = gaussian(),
data = spatial_dat
)
fit_spatial_slope$sdpars$mu
#> spatial(1 | site) spatial(0 + depth | site)
#> 3.808036e-01 4.396188e-06
names(ranef(fit_spatial_slope, "spatial_mu")$terms)
#> [1] "spatial(1 | site)" "spatial(0 + depth | site)"The two SDs answer different biological questions.
spatial(1 | site) is the amount of smooth site-to-site
variation in the expected response after the fixed effects.
spatial(0 + depth | site) is the amount of smooth
site-to-site variation in the depth effect. Large slope SD means the
fitted depth-response gradient changes across the coordinate surface; it
is not a residual correlation and it is not the residual scale
sigma.
Before interpreting the spatial SD or mapped site effects, inspect
the spatial diagnostic row. It is deliberately named
spatial_mu_diagnostics, not
phylo_mu_replication, because the fitted layer is a
coordinate site field:
spatial_check <- check_drm(fit_spatial)
spatial_check[spatial_check$check %in% c(
"optimizer_convergence",
"fixed_gradient",
"hessian_positive_definite",
"spatial_mu_diagnostics"
), ]
#> <drm_check: 4 checks>
#> ok: 4; notes: 0; warnings: 0; errors: 0
#> check status
#> optimizer_convergence ok
#> fixed_gradient ok
#> hessian_positive_definite ok
#> spatial_mu_diagnostics ok
#> value
#> 0
#> max=0.000008559; component=beta_sigma[1]
#> TRUE
#> group=site; n_sites=12; min_site_n=6; coord_range=1.475; spatial_sd=0.3808; sd_ratio=1.214
#> message
#> nlminb convergence code is 0.
#> Maximum absolute fixed gradient is <= 0.001; largest component is beta_sigma[1].
#> sdreport reports a positive-definite Hessian.
#> The coordinate spatial random intercept has replicated sites and a non-negligible fitted SD relative to the residual scale.The spatial row reports the number of fitted sites, the minimum
number of observations per site, the coordinate range used by the fixed
exponential covariance, the fitted spatial SD, and the ratio between the
spatial SD and the residual scale. A note is not a failed
model. It tells you to be cautious when a site is singly observed or
when the spatial field is tiny relative to unstructured residual
variation.
The fitted coords path accepts either one row per site
or one row per observation, provided coordinates are constant within
each site. Internally, the coordinate path builds a fixed exponential
covariance from pairwise site distances, inverts it to a precision
matrix, and estimates one spatial SD for each fitted spatial
coefficient. The intercept model estimates one field. The one-slope
model spatial(1 + depth | site, coords = coords) estimates
two independent fields: spatial(1 | site) for site
intercept deviations and spatial(0 + depth | site) for
site-specific depth slopes. These two fields share the same coordinate
precision but have separate SDs and no intercept-slope
corpair() row. This is a small-data foundation and recovery
target, not the scalable mesh/SPDE implementation. The conditional
spatial effects from ranef() are shrunken site deviations
in the location predictor after the fixed effects and the spatial
covariance have been estimated.
The planned mesh argument plays a different role. A
mesh is the computational scaffold for an SPDE/GMRF field:
a triangular network of vertices where the latent spatial field is
represented and then projected back to observations. Users should not
interpret mesh vertices as biological sampling units; they are numerical
support points for the spatial surface. In other words, a mesh is not
required by the idea of spatial dependence itself. It is required by the
scalable SPDE/GMRF approximation. The friendly API should let users
start with coords = coords; mesh = mesh is the
expert route for reproducible control over boundaries, barriers,
coastlines, or uneven sampling.
The spatial term is deliberately parallel to the phylogenetic term:
# implemented for Gaussian mu/sigma intercepts and matching bivariate blocks
phylo(1 | species, tree = tree)
# implemented for one numeric Gaussian mu slope
phylo(1 + x | species, tree = tree)
# implemented for Gaussian mu/sigma intercepts and matching bivariate blocks
spatial(1 | site, coords = coords)
# implemented for one numeric Gaussian mu slope
spatial(1 + x | site, coords = coords)
# planned
spatial(1 | site, mesh = mesh)The mathematical role is the same: add structured Gaussian
coefficient fields to a distributional predictor. The computational
route differs. phylo() starts from an ultrametric tree with
branch lengths and builds a sparse tree-derived precision. The first
spatial() slice starts from coordinates and builds a fixed
coordinate precision; the future mesh path will build an SPDE/GMRF
precision.
The computational plan is to keep using sparse A-inverse ideas for
phylogeny and add SPDE/GMRF structure for spatial fields. The spatial
route should cite the SPDE/GMRF method literature and the software it
builds on. The key methodological reference is Lindgren, Rue, and
Lindstrom (2011), who connected Gaussian fields to sparse GMRFs through
the SPDE approach (doi:10.1111/j.1467-9868.2011.00777.x).
The ecological TMB precedent is sdmTMB, described in its Journal of
Statistical Software paper as a package for spatial and
spatiotemporal GLMMs using TMB, fmesher, latent GMRFs, and
the SPDE approach. If drmTMB imports fmesher
or asks users to pass fmesher mesh objects, the website and
paper should also tell users to cite fmesher as software.
Any code selectively ported from gllvmTMB,
sdmTMB, fmesher, or INLA-related sources must
be recorded in inst/COPYRIGHTS before the spatial feature
is called complete.
Structured random slopes are staged more conservatively than ordinary
grouped random slopes. Coordinate spatial and phylogenetic Gaussian
mu have first one-slope paths, and q1 structured
sigma one-slope paths fit for phylo/spatial/animal/relmat.
The phylo, A-matrix animal, and K/Q relmat sigma slopes are
inference-ready with caveats; spatial is point-fit/extractor only. These
paths are deliberately independent: each returns separate SDs and no
intercept-slope corpair() row. Additional multiple or
labelled structured-slope layouts outside the exact fitted ledger cells,
mesh/SPDE slopes, and structured slope correlations remain later gates.
The combined phylogeny-plus-spatial route should be a separate
additive-block model with a phylogenetic field and a spatial field, not
one large cross-factor covariance model. A more interesting future
two-response target is the slope1-slope2 correlation for the same
covariate across traits, the plasticity-syndrome question described by
O’Dea, Noble, and Nakagawa (2021).
Meta-analysis and future bivariate structure
In meta-analysis, the separation among known sampling covariance, phylogenetic dependence, and spatial dependence is especially transparent:
The known matrix V handles sampling uncertainty. The
phylogenetic and spatial terms add extra biological or environmental
dependence after that known uncertainty has been included. The same
design will let drmTMB support structured Gaussian
meta-analysis while keeping ecological and evolutionary meta-analyses
readable.
The first phylogenetic target is the location-scale model. In
bivariate Gaussian models, matching mu1/mu2
phylogenetic location terms estimate the mean-mean phylogenetic
correlation. Matching labelled phylo() terms across
mu1, mu2, sigma1, and
sigma2 now fit the first constant q=4 phylogenetic
location-scale block: one location-location row, four location-scale
rows, and one scale-scale row in
corpairs(level = "phylogenetic"). The six q=4 correlations
are latent phylogenetic random-effect correlations, not residual
rho12.
The first fitted bivariate phylogenetic location-coscale syntax looks like this:
fit_biv_phylo <- drmTMB(
formula = drm_formula(
mu1 = log_body_mass ~ lifestyle + phylo(1 | species, tree = tree),
mu2 = log_litter_size ~ lifestyle + phylo(1 | species, tree = tree),
sigma1 = ~ lifestyle,
sigma2 = ~ lifestyle,
rho12 = ~ lifestyle
),
family = c(gaussian(), gaussian()),
data = mammals
)This syntax fits correlated phylogenetic random intercepts in
mu1 and mu2. It estimates two phylogenetic
location SDs and one phylogenetic mean-mean correlation. The
sigma1, sigma2, and residual
rho12 formulas remain ordinary fixed-effect distributional
parameters in this first slice. Use
phylo(1 | p | species, tree = tree) in both location
formulas when you want the fitted phylogenetic mean-mean row to use a
block label such as p.
The Box 1 direct-SD alternative for two responses keeps the phylogenetic effects in the location formulas, but lets species-level predictors explain the magnitude of each response’s phylogenetic location variation:
fit_biv_sd_phylo <- drmTMB(
formula = drm_formula(
mu1 = heat_tolerance ~ climate +
phylo(1 | species, tree = tree),
mu2 = desiccation_tolerance ~ climate +
phylo(1 | species, tree = tree),
sigma1 = ~ habitat_variability,
sigma2 = ~ habitat_variability,
rho12 = ~ assay_context,
sd_phylo1(species) ~ habitat_variability_species,
sd_phylo2(species) ~ habitat_variability_species
),
family = c(gaussian(), gaussian()),
data = tolerance
)Here sd_phylo1(species) targets the mu1
phylogenetic location SD surface and sd_phylo2(species)
targets the mu2 surface. The right-hand-side predictors
must be constant within species after model-row filtering. The latent
phylogenetic mean-mean correlation remains a constant fitted
correlation, and rho12 remains the within-observation
residual correlation:
The corpairs() row is the fitted phylogenetic
correlation point estimate between the two location random effects; this
direct-SD route does not have validated interval evidence. The
rho12() values are residual correlations between the two
responses after the fitted fixed effects and phylogenetic effects have
been included. Run check_drm(fit_biv_sd_phylo) before
interpreting the direct-SD slopes; the diagnostic reports species
replication, each fitted species-SD range, and whether any fitted
direct-SD surface is non-positive or non-finite.
Mathematically, the direct-SD model replaces constant phylogenetic location SDs with species-level SD surfaces. For response :
The current bivariate direct-SD path keeps the cross-response phylogenetic location correlation constant:
This is why
sd_phylo1(species) ~ habitat_variability_species and
sd_phylo2(species) ~ habitat_variability_species answer a
different question from the q=4 location-scale block. They ask whether
species-level predictors change the magnitude of phylogenetic mean
variation for each response. They do not put random effects into
sigma1 or sigma2.
The full constant q=4 location-scale form uses an explicit covariance-block label:
fit_biv_phylo_q4 <- drmTMB(
formula = drm_formula(
mu1 = heat_tolerance ~ climate + phylo(1 | p | species, tree = tree),
mu2 = desiccation_tolerance ~ climate +
phylo(1 | p | species, tree = tree),
sigma1 = ~ habitat_variability +
phylo(1 | p | species, tree = tree),
sigma2 = ~ habitat_variability +
phylo(1 | p | species, tree = tree),
rho12 = ~ assay_context
),
family = c(gaussian(), gaussian()),
data = tolerance
)
corpairs(fit_biv_phylo_q4, level = "phylogenetic")
rho12(fit_biv_phylo_q4)Partial q=4 syntax, unlabelled all-four syntax, random slopes, and
multiple phylogenetic blocks remain outside this first fitted path. The
q=4 corpairs() output has six phylogenetic rows: one
location-location row, four location-scale rows, and one scale-scale
row. The four location-scale rows include both same-trait and
cross-trait mean-scale pairs. These rows are the latent phylogenetic
correlations among species-level effects. They are not the residual
rho12 curve, which is why the example shows
corpairs() and rho12() separately.
For the simpler fitted mean-mean phylogenetic correlation point
estimate, use corpars$phylo,
corpairs(fit_biv_phylo, level = "phylogenetic"), or
summary(fit_biv_phylo)$covariance. Run
check_drm(fit_biv_phylo) before interpreting that row: the
current diagnostic flags phylogenetic correlations near the boundary and
phylogenetic location SDs that are tiny relative to the matching
residual scales. profile_targets() can expose a callable
diagnostic profile target, but that does not validate an interval or
promote this intercept-only q2 row. The q=4 correlations are derived
from theta_phylo, so direct profile intervals for those six
rows are not yet claimed:
phylo_target <- "cor:phylo:cor(mu1:(Intercept),mu2:(Intercept) | phylo | species)"
corpairs(fit_biv_phylo, level = "phylogenetic")
summary(fit_biv_phylo)$covariance
profile_targets(fit_biv_phylo)Do not read a phylogenetic species effect as a residual
observation-level correlation. The phylo() layer is
species-level structure induced by the tree; residual rho12
is within-observation coupling between the two measured responses. A
separate non-phylogenetic species-level block can be useful in a
specialized model, but it is an advanced comparison because it tries to
separate two species-level covariance layers that use the same grouping
factor. That comparison should live in a dedicated identifiability
example, not in this introductory ladder.
The residual rho12 term remains distinct from the
phylogenetic or spatial correlation matrices. rho12 is the
residual coupling between two responses at the observation level;
A and M describe dependence among species or
places. In future bivariate structural models, drmTMB
should also estimate level-specific correlations such as phylogenetic
mean-mean correlations, non-phylogenetic species correlations, spatial
field correlations, and mean-scale correlations. These are not residual
rho12; they should be reported as structural or group-level
covariance summaries. For the univariate structural-dependence ladder,
keep the implementation status visible: use
animal(pedigree/A/Ainv = ...) for the animal-model first
slice, use phylo() when the species tree is the fitted
dependence structure, use spatial() when the site field is
the fitted dependence structure, plan combined phylo() plus
spatial() only after joint identifiability checks, and use
relmat(K/Q = ...) for lower-level known-matrix latent
random effects.