When variance carries signal, Part 1: location-scale models
Source:vignettes/location-scale.Rmd
location-scale.RmdThis tutorial starts with a biological question: do two habitats
differ only in mean growth, or does one habitat also make growth less
predictable? In a Gaussian location-scale model, the location part
models the expected response mu, while the scale part
models the residual standard deviation sigma. That residual
scale is not just a nuisance parameter; it can be the scientific answer
when the question is about individual variability, predictability, or
remaining heterogeneity after the mean model is accounted for.
This is Part 1 of a two-article sequence. It models the response mean
mu and the residual standard deviation sigma.
Continue to Part 2:
location-scale-scale models when a predictor should model the
standard deviation of a latent group or phylogenetic random effect
through sd().
Read this after you have fit a first model in Distributional regression with drmTMB and
confirmed Gaussian is the right family in Choosing response families; this
tutorial goes deeper into interpreting mu and
sigma once that choice is made.
If you are fitting your first model, start with the worked growth example, then return to the syntax overview below when you want to adapt the formula.
The core Gaussian location-scale route supports fixed effects,
optional random effects in the location formula, and residual-scale
random intercepts and independent random slopes in the
sigma formula. The same pattern is used throughout the
documentation: write the model symbolically first, show the matching R
syntax, fit the model, and interpret the fitted output.
Model equations and matching R syntax
For Gaussian location-scale regression, each estimated parameter has its own linear predictor:
The matching syntax is:
The location formula defines the design matrix for beta;
the scale formula defines the design matrix for gamma. The
log link keeps the fitted residual SD positive.
| Quantity | R source | Interpretation |
|---|---|---|
mu_i |
y ~ x |
expected response |
beta |
coef(fit, "mu") |
additive effects on the expected response |
sigma_i |
sigma ~ z |
residual SD around the fitted mean |
gamma |
coef(fit, "sigma") |
effects on log residual SD |
Grouped effects do not change the meaning of sigma
A random intercept in the location model adds among-group variation in expected responses:
Here sd_mu_group is the SD of group-level mean
deviations. It is not the residual SD sigma. A random
effect inside the sigma formula asks a third question: do
groups differ in their residual variability?
More elaborate ordinary intercept-slope blocks and their covariance output are documented in Which scale are you modelling? and Structural dependence. To model predictors of the group-level SD itself, continue to Part 2: location-scale-scale models.
Interpret sigma on the SD or variance scale
Because log(sigma_i) = z_i^T gamma, exponentiating a
scale coefficient gives an SD ratio. If
gamma_temperature = 0.3, then a one-unit temperature
increase multiplies residual SD by exp(0.3) = 1.35.
Squaring the ratio gives the residual-variance ratio,
exp(2 * 0.3) = 1.82.
| Formula component | Report | Biological reading |
|---|---|---|
growth ~ temperature |
beta_temperature |
additive change in expected growth |
sigma ~ temperature |
exp(gamma_temperature) |
residual-SD ratio per temperature unit |
(0 + temperature | population) in mu
|
random-slope SD | variation among population-specific mean slopes |
sigma ~ (0 + temperature | population) |
residual-scale random-slope SD | variation among population-specific log-SD slopes |
Name the measured response and model component before stating a
biological conclusion. “Temperature-dependent variability” is ambiguous
until the reader knows whether the model changed residual
sigma, a mean random-slope SD, or a random effect inside
sigma.
Worked example: growth mean and predictability
Suppose an ecologist measures juvenile growth in two habitats and records the temperature at each observation. The location question is whether mean growth differs between habitats and changes with temperature. The scale question is whether residual growth variability differs between habitats after accounting for those mean effects.
For this example, the fitted model is:
The matching drmTMB syntax is:
drmTMB(
drm_formula(growth ~ habitat + temperature, sigma ~ habitat),
family = gaussian(),
data = dat
)The code below simulates one dataset with the same structure. Simulated data keep the vignette small and reproducible; a real analysis would replace this block with field or laboratory measurements.
set.seed(42)
n <- 240
dat <- data.frame(
habitat = factor(rep(c("forest", "grassland"), each = n / 2)),
temperature = rnorm(n)
)
mu <- 8 + 1.2 * (dat$habitat == "grassland") + 0.6 * dat$temperature
sigma_true <- exp(log(0.6) + 0.55 * (dat$habitat == "grassland"))
dat$growth <- rnorm(n, mean = mu, sd = sigma_true)Fit the location-scale model:
fit_growth <- drmTMB(
drm_formula(growth ~ habitat + temperature, sigma ~ habitat),
family = gaussian(),
data = dat
)Before interpreting coefficients, check the fitted object:
check_drm(fit_growth)
#> <drm_check: 12 checks>
#> ok: 12; notes: 0; warnings: 0; errors: 0
#> check status
#> optimizer_convergence ok
#> optimizer_budget ok
#> finite_objective ok
#> logsigma_clamp_active ok
#> fixed_gradient ok
#> sdreport_status ok
#> hessian_positive_definite ok
#> standard_errors_finite ok
#> standard_errors_inflated ok
#> dropped_rows ok
#> positive_scale ok
#> fixed_effect_design_size ok
#> value
#> 0
#> iterations=19; function=33; gradient=19
#> 279.3
#> <NA>
#> max=0.0008192; component=beta_sigma[2]
#> ok
#> TRUE
#> range=[0.04551,0.1102]
#> n_inflated=0; max_se=0.1102; median_se=0.06466
#> nobs=240; dropped=0
#> min=0.5633
#> total_mb=0.04107; max_cols=3; largest=mu; largest_class=matrix; largest_density=0.8333
#> message
#> nlminb convergence code is 0.
#> Optimizer evaluation counts recorded; no eval.max or iter.max control was supplied.
#> Objective and log-likelihood are finite.
#> The log(sigma) clamp is not active at the optimum.
#> Maximum absolute fixed gradient is <= 0.001; largest component is beta_sigma[2].
#> TMB::sdreport() completed successfully.
#> sdreport reports a positive-definite Hessian.
#> All fixed-effect standard errors are finite.
#> No fixed-effect standard error is inflated relative to the others.
#> No rows were dropped by model-frame or known-covariance filtering.
#> All fitted scale values are finite and positive.
#> Dense fixed-effect design matrices are modest for this fit.The interval target inventory is part of the same interpretation gate. For this fixed-effect example, every coefficient is a direct profile target:
profile_targets(fit_growth)[
,
c("parm", "estimate", "profile_ready", "profile_note")
]
#> parm estimate profile_ready profile_note
#> 1 fixef:mu:(Intercept) 7.9948246 TRUE ready
#> 2 fixef:mu:habitatgrassland 1.2068575 TRUE ready
#> 3 fixef:mu:temperature 0.5745448 TRUE ready
#> 4 fixef:sigma:(Intercept) -0.5740134 TRUE ready
#> 5 fixef:sigma:habitatgrassland 0.6376342 TRUE readyNow print the coefficient table:
summary(fit_growth)
#> <summary.drmTMB>
#> estimator: ML
#> estimate std_error
#> mu:(Intercept) 7.9948246 0.05143610
#> mu:habitatgrassland 1.2068575 0.11021142
#> mu:temperature 0.5745448 0.04550947
#> sigma:(Intercept) -0.5740134 0.06466108
#> sigma:habitatgrassland 0.6376342 0.09160089
#> Distributional, random-effect, scale, and correlation parameters:
#> component dpar term estimate minimum
#> fitted:sigma distributional-scale sigma fitted range 0.8144743 0.5632603
#> maximum scale
#> fitted:sigma 1.065688 response
#> logLik: -279.3
#> convergence: 0How to read this output:
- Rows beginning with
mu:are mean-growth effects on the response scale. For example,mu:temperatureis the expected change in mean growth for a one-unit increase in temperature. - Rows beginning with
sigma:are log-residual-standard-deviation effects. For example,sigma:habitatgrasslandis not an additive change in growth; it is a log-scale change in residual variability. - Exponentiating a
sigmacoefficient gives the multiplicative change in residual standard deviation.
sigma_habitat <- coef(fit_growth, "sigma")["habitatgrassland"]
data.frame(
coefficient = sigma_habitat,
residual_sd_ratio = exp(sigma_habitat),
residual_variance_ratio = exp(2 * sigma_habitat)
)
#> coefficient residual_sd_ratio residual_variance_ratio
#> habitatgrassland 0.6376342 1.892 3.579662In this fitted example, exp(sigma:habitatgrassland) is
the estimated ratio of grassland residual SD to forest residual SD. A
value near 2 means that grassland has about twice the residual SD of
forest after accounting for mean habitat and temperature effects.
Because residual variance is SD squared, the same fitted coefficient
implies about four times the residual variance.
It is often clearer to report the fitted residual SDs and variances directly:
newdat <- data.frame(
habitat = factor(c("forest", "grassland"), levels = levels(dat$habitat)),
temperature = 0
)
growth_report <- data.frame(
habitat = newdat$habitat,
fitted_mean_growth = predict(fit_growth, newdata = newdat, dpar = "mu"),
fitted_residual_sd = predict(fit_growth, newdata = newdat, dpar = "sigma")
)
growth_report$fitted_residual_variance <- growth_report$fitted_residual_sd^2
growth_report
#> habitat fitted_mean_growth fitted_residual_sd fitted_residual_variance
#> 1 forest 7.994825 0.5632603 0.3172622
#> 2 grassland 9.201682 1.0656882 1.1356914This table maps the model back to the scientific question. The mean column summarises the location model. The residual SD column summarises the scale model in the units of growth. The residual variance column is the variance-facing version of the same fitted Gaussian scale model.
The fitted mean can be checked against the raw response pattern. Here
the points are observed growth values; the fitted lines and ribbons come
from predict_parameters() on an explicit
temperature-by-habitat grid:
growth_mu_grid <- prediction_grid(
fit_growth,
focal = c("temperature", "habitat"),
at = list(
temperature = seq(
min(dat$temperature),
max(dat$temperature),
length.out = 80
)
)
)
growth_mu_surface <- predict_parameters(
fit_growth,
newdata = growth_mu_grid,
dpar = "mu",
conf.int = TRUE
)
unique(growth_mu_surface[, c(
"dpar",
"conf.status",
"interval_source",
"conf.level"
)])
#> dpar conf.status interval_source conf.level
#> 1 mu wald wald 0.95
if (requireNamespace("ggplot2", quietly = TRUE)) {
ggplot2::ggplot(
dat,
ggplot2::aes(x = temperature, y = growth, colour = habitat)
) +
ggplot2::geom_point(alpha = 0.38, size = 1.25) +
ggplot2::geom_ribbon(
data = growth_mu_surface,
ggplot2::aes(
x = temperature,
ymin = conf.low,
ymax = conf.high,
fill = habitat
),
inherit.aes = FALSE,
alpha = 0.18,
colour = NA
) +
ggplot2::geom_line(
data = growth_mu_surface,
ggplot2::aes(y = estimate),
linewidth = 0.85
) +
location_scale_habitat_scales() +
ggplot2::labs(
title = "Mean growth and observed scatter",
subtitle = "Points are raw growth; ribbons are 95% Wald bands for mu",
x = "Temperature",
y = "Growth",
colour = "Habitat",
fill = "Habitat"
) +
location_scale_theme() +
ggplot2::guides(fill = "none")
}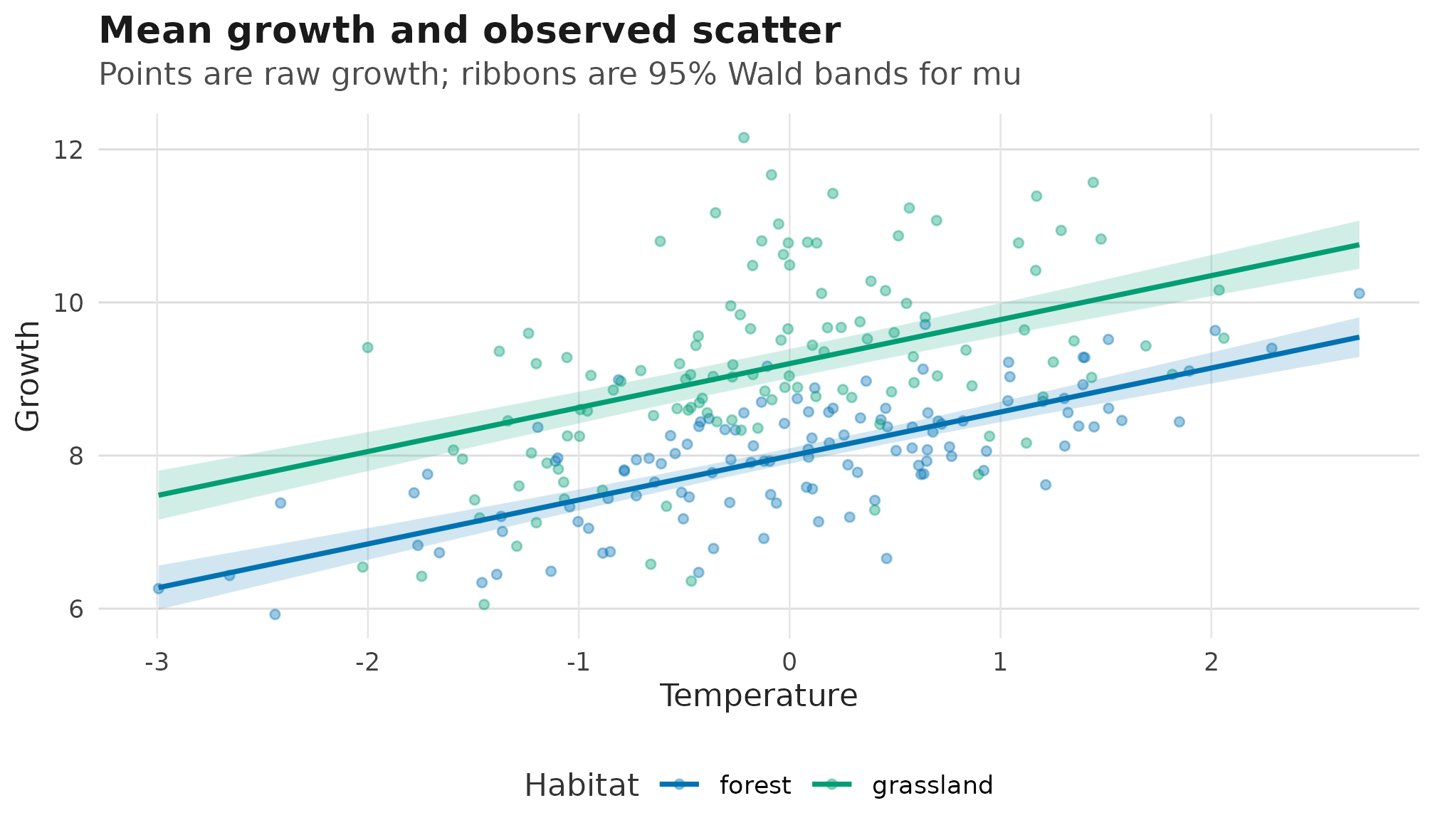
Raw growth observations and fitted response-scale mu
surfaces for the Gaussian location-scale example; ribbons are 95% Wald
confidence bands from predict_parameters().
The scale model is a different display. Do not put raw
growth points on the sigma axis; plot the
fitted residual SD and name the interval source:
growth_sigma_grid <- prediction_grid(
fit_growth,
focal = "habitat",
condition = list(temperature = 0)
)
growth_sigma_surface <- predict_parameters(
fit_growth,
newdata = growth_sigma_grid,
dpar = "sigma",
conf.int = TRUE
)
unique(growth_sigma_surface[, c(
"dpar",
"conf.status",
"interval_source",
"conf.level"
)])
#> dpar conf.status interval_source conf.level
#> 1 sigma wald wald 0.95
if (requireNamespace("ggplot2", quietly = TRUE)) {
ggplot2::ggplot(
growth_sigma_surface,
ggplot2::aes(x = estimate, y = habitat, colour = habitat)
) +
ggplot2::geom_segment(
ggplot2::aes(x = conf.low, xend = conf.high, yend = habitat),
linewidth = 0.8
) +
ggplot2::geom_point(size = 2.8) +
location_scale_habitat_scales() +
ggplot2::labs(
title = "Residual SD differs by habitat",
subtitle = "Points are fitted sigma values; bars are 95% Wald intervals",
x = "Fitted residual SD (sigma)",
y = NULL,
colour = "Habitat"
) +
location_scale_theme() +
ggplot2::guides(colour = "none")
}
#> Warning: No shared levels found between `names(values)` of the manual scale and the
#> data's fill values.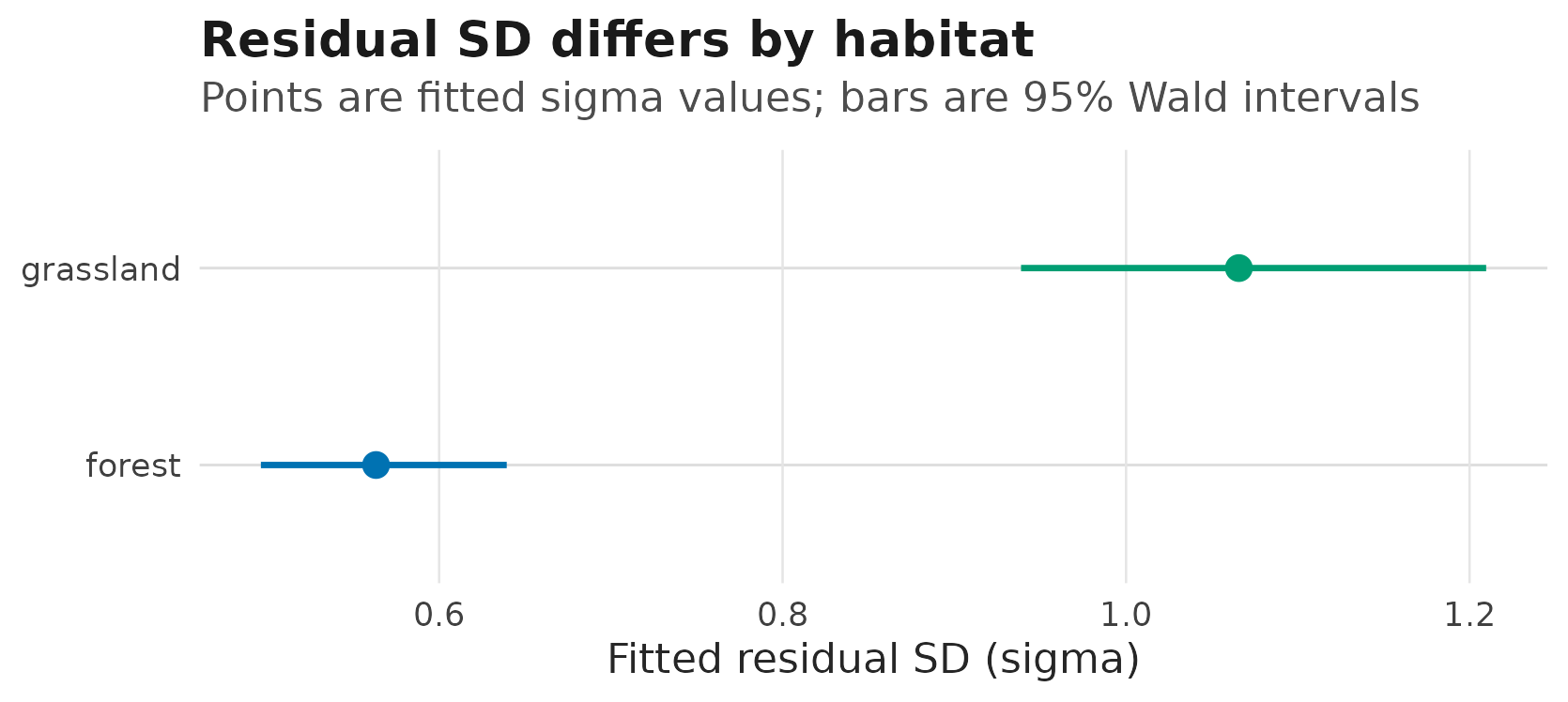
Fitted residual standard deviations by habitat at average temperature;
horizontal intervals are 95% Wald confidence intervals requested from
predict_parameters().
The compact translation table below is the reporting layer Pat should be able to read without returning to the equations:
growth_translation <- data.frame(
model_piece = c(
"fixed mean slope",
"fixed residual-SD contrast",
"fixed residual-variance contrast"
),
fitted_term = c(
"mu:temperature",
"exp(sigma:habitatgrassland)",
"exp(2 * sigma:habitatgrassland)"
),
response_scale_value = c(
unname(coef(fit_growth, "mu")["temperature"]),
unname(exp(sigma_habitat)),
unname(exp(2 * sigma_habitat))
),
interpretation = c(
"mean growth change per one-unit temperature increase",
"grassland residual SD divided by forest residual SD",
"grassland residual variance divided by forest residual variance"
)
)
growth_translation$response_scale_value <-
round(growth_translation$response_scale_value, 3)
growth_translation
#> model_piece fitted_term
#> 1 fixed mean slope mu:temperature
#> 2 fixed residual-SD contrast exp(sigma:habitatgrassland)
#> 3 fixed residual-variance contrast exp(2 * sigma:habitatgrassland)
#> response_scale_value
#> 1 0.575
#> 2 1.892
#> 3 3.580
#> interpretation
#> 1 mean growth change per one-unit temperature increase
#> 2 grassland residual SD divided by forest residual SD
#> 3 grassland residual variance divided by forest residual varianceA report can now say three concrete things. Mean growth increases by
the fitted mu:temperature slope for each one-unit
temperature increase. Grassland has the fitted residual-SD ratio in the
second row after accounting for mean habitat and temperature effects.
Because Gaussian variance is sigma^2, the residual variance
contrast is the third row. If that third row is larger than one,
individual growth is less predictable in grassland.
The same reporting discipline carries over to hierarchical versions of the question:
| If the biological question is… | Fit this kind of term | Report this quantity | Check before reporting |
|---|---|---|---|
| Do populations differ in thermal plasticity? |
(0 + temperature | population) or
(1 + temperature | population) in the mu
formula |
the random-slope SD, in growth-per-temperature units |
check_drm(fit) and the matching
sd:mu:...temperature... row in
profile_targets(fit)
|
| Do high-baseline populations also have steeper thermal reaction norms? |
(1 + temperature | population) in the mu
formula |
the random-slope SD, and when the correlated block is fitted, the
group-level intercept-slope correlation from
corpairs(fit, class = "mean-slope")
|
check_drm(fit), the matching
sd:mu:...temperature... row, and the fitted correlation row
in profile_targets(fit)
|
| Does predictability change with temperature? | sigma ~ temperature |
exp(gamma_temperature) as the residual-SD ratio, and
exp(2 * gamma_temperature) when the paper talks about
variance |
check_drm(fit) and the
fixef:sigma:temperature row in
profile_targets(fit)
|
| Do habitats differ in among-population variation? |
(1 | population) plus
sd(population) ~ habitat
|
exp(alpha_habitat) as the ratio of among-population
SDs |
check_drm(fit) and the
fixef:sd(population):... row in
profile_targets(fit)
|
These three rows answer different biological questions.
sigma ~ temperature models residual variation among
observations. (0 + temperature | population) models
population-to-population differences in the mean slope.
sd(population) ~ habitat models the size of
among-population mean differences.
Curved responses and interactions
Quadratic terms and interactions can appear in either formula:
fit_curve <- drmTMB(
bf(
growth ~ habitat * temperature + I(temperature^2),
sigma ~ I(temperature^2)
),
family = gaussian(),
data = dat
)A linear coefficient is only a local slope when its model also contains a quadratic term or interaction. Interpret the fitted curve at biologically meaningful predictor values rather than reading that coefficient alone:
curve_grid <- expand.grid(
habitat = c("forest", "grassland"),
temperature = c(-1.5, 0, 1.5)
)
data.frame(
curve_grid,
mu = predict(fit_curve, newdata = curve_grid, dpar = "mu"),
sigma = predict(fit_curve, newdata = curve_grid, dpar = "sigma")
)If the sigma formula is omitted, drmTMB
fits one intercept-only residual SD. Use that simpler model only when a
common residual SD is scientifically and diagnostically adequate.
Where to continue
- Use Part 2: location-scale-scale
models when predictors model a grouped or phylogenetic random-effect
SD through
sd(). - Use Mean effects and residual
heterogeneity for Gaussian meta-analysis with known sampling
covariance through
meta_V(V = V). - Use Changing residual coupling with rho12 for two responses and predictor-dependent residual correlation.
- Use Structural dependence
for
phylo(),animal(),spatial(), andrelmat().
Current implementation boundaries
- This article documents the univariate Gaussian
muandsigmaroute. - Random intercepts, independent slopes, and ordinary correlated
intercept-slope blocks are available in
mu;sigmasupports its documented random-intercept and random-slope slices. - Scale coefficients use a log-SD link. Exponentiate once for an SD ratio and twice in the exponent for a variance ratio.
- Missing rows are removed when variables used by either formula are missing.
- A fitted optimizer code is not enough: run
check_drm(), inspect gradient and Hessian diagnostics, and use What can I trust? before widening the claim. -
sigmais not a group random-effect SD. Use Part 2 for the separatesd(group) ~ predictorsgrammar.