Individual morphometrics: the simplest GLLVM
Source:vignettes/articles/morphometrics.Rmd
morphometrics.RmdThis is the simplest case the package handles, and it is the foundation for everything else on the homepage guide. One observational level (individuals), several continuous traits per individual, and shared low-rank covariance between traits. No phylogeny, no spatial structure, no nesting. This article walks through that model end-to-end.
The recovery numbers and intervals below describe this teaching example; they do not establish calibrated coverage across other sample sizes or model structures.
The working question is simple: if five body measurements are all partly driven by size and shape, can a rank-2 GLLVM recover that shared structure and still leave trait-specific variation on the diagonal?
Example Data
We use a prepared example object rather than asking you to read the
data-generating script first. The script is still in the repository
(data-raw/examples/make-morphometrics-example.R) so the
fixture is reproducible, but the article starts where a user starts:
with data, truth, formulas, and one biological question.
morph <- readRDS(system.file(
"extdata", "examples", "morphometrics-example.rds",
package = "gllvmTMB"
))
df <- morph$data_long
df_wide <- morph$data_wide
truth <- morph$truth
estimands <- morph$estimands
morph$story$question
#> [1] "Do five body measurements share two latent axes, size and shape, while retaining trait-specific variation?"
head(df, 8)
#> individual trait value
#> 1 1 length -0.04447039
#> 2 1 mass -1.23031062
#> 3 1 wing 0.23493689
#> 4 1 tarsus 0.64897692
#> 5 1 bill -0.55819209
#> 6 2 length 0.06037967
#> 7 2 mass 0.43283075
#> 8 2 wing 0.48482650The object represents 150 individual birds measured on five traits: body length, mass, wing chord, tarsus length, and bill depth. The true loading matrix has a size axis (factor 1, all positive: bigger birds tend to be bigger on every measurement) and a shape axis (factor 2, mixed signs: long-and-skinny vs. short-and-stocky). Each trait also has a trait-specific diagonal variance .
round(Sigma_true, 2)
#> length mass wing tarsus bill
#> length 0.97 0.92 0.68 0.58 0.60
#> mass 0.92 1.14 0.72 0.60 0.72
#> wing 0.68 0.72 1.00 0.76 0.24
#> tarsus 0.58 0.60 0.76 0.86 0.12
#> bill 0.60 0.72 0.24 0.12 0.90
estimands
#> trait shared_variance unique_variance total_variance communality
#> 1 length 0.82 0.15 0.97 0.8453608
#> 2 mass 1.04 0.10 1.14 0.9122807
#> 3 wing 0.80 0.20 1.00 0.8000000
#> 4 tarsus 0.74 0.12 0.86 0.8604651
#> 5 bill 0.72 0.18 0.90 0.8000000Pitfall: factor level order. The example object pins
levels(df$trait)totruth$trait_names. Without explicit levels, R may sort levels alphabetically (bill, length, mass, tarsus, wing), and the rows of fitted will not match the rows of the truth table. The model is correct; the comparison is not.
Fit
The same model can be fitted from long or wide data. The long form is
canonical because it makes the (individual, trait) rows
explicit; the wide forms are the convenient equivalents for readers who
already have one row per individual and one column per trait.
# The object stores the exact long and wide formulas used below:
morph$formula_long
#> value ~ 0 + trait + latent(0 + trait | individual, d = 2)
morph$formula_wide
#> traits(length, mass, wing, tarsus, bill) ~ 1 + latent(1 | individual,
#> d = 2)
# Long format (canonical)
fit_long <- gllvmTMB(
morph$formula_long,
data = df,
trait = morph$fit_args$trait,
unit = morph$fit_args$unit,
family = morph$fit_args$family
)
# Wide data-frame format (same model with compact formula syntax)
fit_wide_formula <- gllvmTMB(
morph$formula_wide,
data = df_wide,
unit = morph$fit_args$unit,
family = morph$fit_args$family
)
# The rest of the article uses the canonical long-format fit.
fit <- fit_long
as.numeric(logLik(fit))
#> [1] -739.6107
cat("Long/formula-wide logLik difference:",
signif(abs(as.numeric(logLik(fit_long)) -
as.numeric(logLik(fit_wide_formula))), 3),
"\n")
#> Long/formula-wide logLik difference: 0gllvmTMB defaults to unit = "site", meaning it looks for
a column literally named site in the data. This
morphometric data set has no site column — the grouping
factor is individual. Passing
unit = "individual" redirects the engine to the correct
column; everything else is standard GLLVM machinery.
traits(length, mass, wing, tarsus, bill) is the
formula-level wide data-frame marker. Because the response traits are
named on the LHS, the RHS can use the compact shorthand: 1
becomes the trait-specific intercepts 0 + trait, and
latent(1 | individual) becomes
latent(0 + trait | individual).
What model did we fit?
For each individual we observe a vector of trait measurements — for example, body length, mass, wing chord, tarsus, bill depth in a sparrow study; tail length, fin span, gill area in a fish study; or six items on a psychometric scale in a behavioural study. The reduced-rank GLLVM is
where
is the loading vector for trait
,
is a
-dimensional
latent factor for individual
,
and
is the trait-specific residual with variance
.
Ordinary latent() carries those residual variances on the
diagonal of
by default. The implied between-individual trait covariance is
For traits and factors this is still a small example; the practical gain is interpretability, not a dramatic parameter saving. As grows, the low-rank part scales with loadings rather than the full covariance surface. That low-rank structure is the point of GLLVMs: shared biological axes first, trait-specific variation second.
Diagonal with cross-sectional data. With cross-sectional data (one observation per individual per trait), the
(trait, individual)pair uniquely identifies every row. gllvmTMB detects this automatically: it fixes to a negligibly small constant (and prints ani Auto-suppressing sigma_eps: ...message), so the diagonal term absorbs the full observation-level residual and becomes the trait-specific residual variance . The confounding that would arise if both and were estimated freely is therefore resolved by construction — you do not need to add a separate diagonal term for this default latent model. With longitudinal data (multiple sessions per individual), both terms are separately identified and can be estimated without any auto-suppression; the diagonal term then captures genuine between-individual trait-specific variance on top of the latent structure. See the Covariance & correlation article for the side-by-side comparison.
# Trait-specific unique variances (psi_t)
psi_hat <- extract_Sigma(fit, level = "unit", part = "unique")$s
cat("Estimated psi_t:", round(psi_hat, 3), "\n")
#> Estimated psi_t: 0.129 0.087 0.223 0.132 0.205
cat("True psi_t:", round(psi_true, 3), "\n")
#> True psi_t: 0.15 0.1 0.2 0.12 0.18The extractor stores the diagonal entries in a vector named
s. In the text we call the same entries psi_t,
and the diagonal matrix they form is Psi.
Recovered vs truth
The numbers below describe this one simulated draw of 150 individuals — descriptive recovery on a single dataset, not repeated sampling (see the scope note further down).
Sigma_hat <- extract_Sigma(fit, level = "unit")$Sigma
round(Sigma_hat, 2)
#> length mass wing tarsus bill
#> length 1.18 1.13 0.87 0.77 0.67
#> mass 1.13 1.33 0.86 0.74 0.80
#> wing 0.87 0.86 1.14 0.89 0.30
#> tarsus 0.77 0.74 0.89 1.00 0.18
#> bill 0.67 0.80 0.30 0.18 0.93
cat("Frobenius |Sigma_hat - Sigma_true| / |Sigma_true| =",
round(norm(Sigma_hat - Sigma_true, "F") / norm(Sigma_true, "F"), 3), "\n")
#> Frobenius |Sigma_hat - Sigma_true| / |Sigma_true| = 0.197A row-level comparison shows whether the recovered trait correlations agree with truth. Zero means the fitted correlation equals the known simulation truth for that trait pair:
rownames(Sigma_true) <- colnames(Sigma_true) <- trait_names
rownames(Sigma_hat) <- colnames(Sigma_hat) <- trait_names
corr_compare <- compare_Sigma_table(
fit,
truth = Sigma_true,
level = "unit",
measure = "correlation",
entries = "upper"
)
plot_Sigma_comparison(corr_compare, measure = "correlation")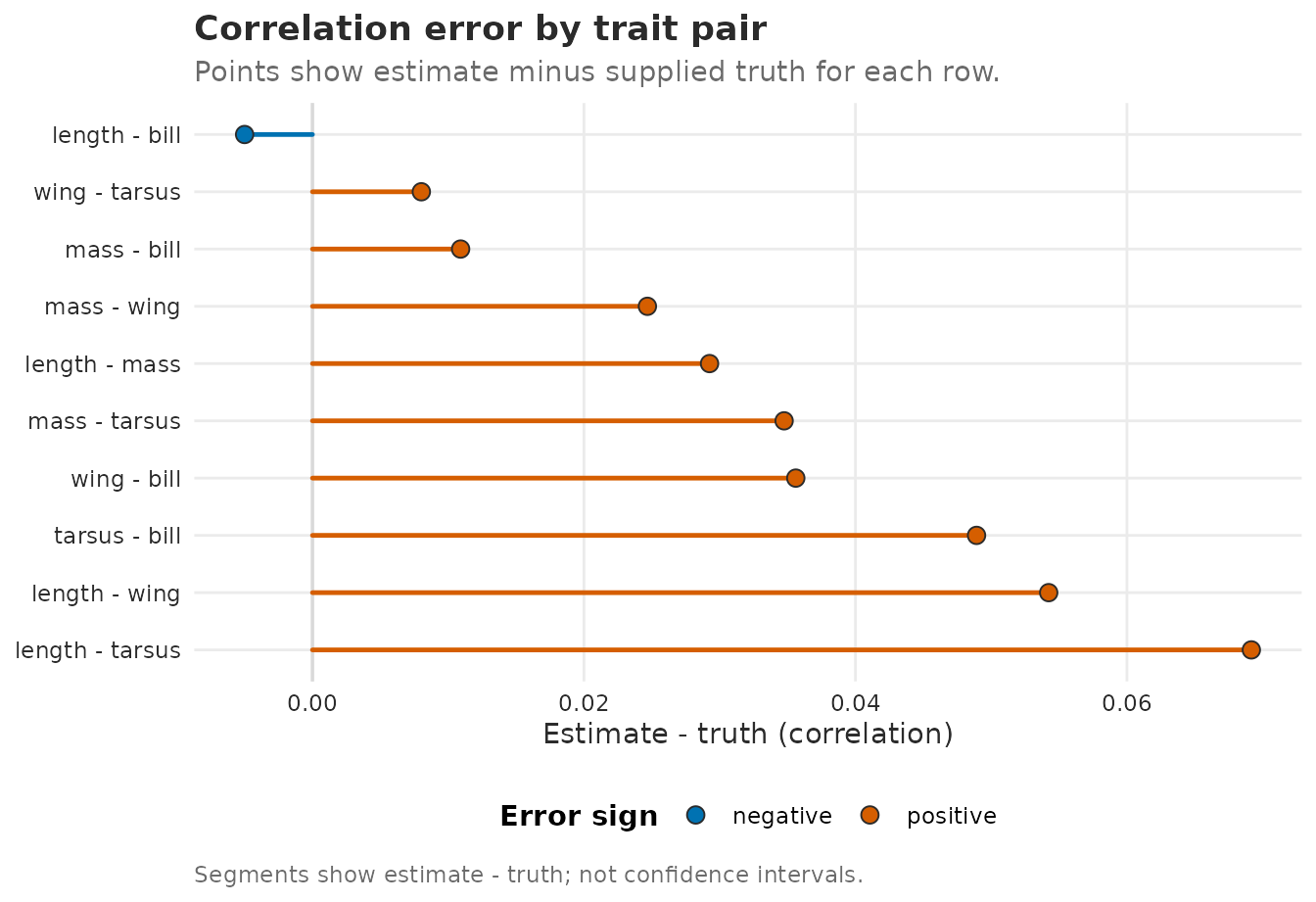
Trait-pair correlation errors from the total between-unit covariance Sigma_B = Lambda Lambda^T + Psi. Points show fitted minus true correlation; zero means exact recovery for that pair.
What
extract_Sigma(level = "unit")$Sigmareturns. The total between-unit covariance — the sum of the reduced-rank shared component and the diagonal unique component. If you requestlatent(..., unique = FALSE), and you get only , which understates the diagonal for this data-generating process. The defaultlatent()fit above includes . Usepart = "shared"/part = "unique"to pull just one component.
Pairwise correlations and communality
For inference on individual cross-trait correlations or per-trait
communality, the canonical extractors return tidy data frames.
extract_correlations() defaults to point estimates.
Fisher-z bounds are an opt-in sensitivity display, not a calibrated
mixed-model interval. The table below therefore reports the fitted
correlations without implying universal coverage. The table is useful
for reporting exact numbers; the plot shows which trait pairs have the
strongest evidence of positive or negative association. Communality
point estimates are enough for this first worked example. When interval
estimates matter, prefer profile or Wald-style intervals where the
target supports them. Bootstrap is a slower, deliberate option for a
separate cached check. This article loads a small cached
bootstrap_Sigma() object for the correlation figure so
pkgdown does not refit bootstrap replicates while rendering.
corr_rows <- extract_correlations(fit, tier = "unit")
corr_rows
#> tier trait_i trait_j correlation lower upper method interval_status
#> 1 B length mass 0.9041350 NA NA none none
#> 2 B length wing 0.7446723 NA NA none none
#> 3 B mass wing 0.6990173 NA NA none none
#> 4 B length tarsus 0.7041940 NA NA none none
#> 5 B mass tarsus 0.6407146 NA NA none none
#> 6 B wing tarsus 0.8275512 NA NA none none
#> 7 B length bill 0.6371630 NA NA none none
#> 8 B mass bill 0.7217348 NA NA none none
#> 9 B wing bill 0.2885881 NA NA none none
#> 10 B tarsus bill 0.1853213 NA NA none none
plot_correlations(morph_boot_R, tier = "unit", style = "eye", sort = "trait")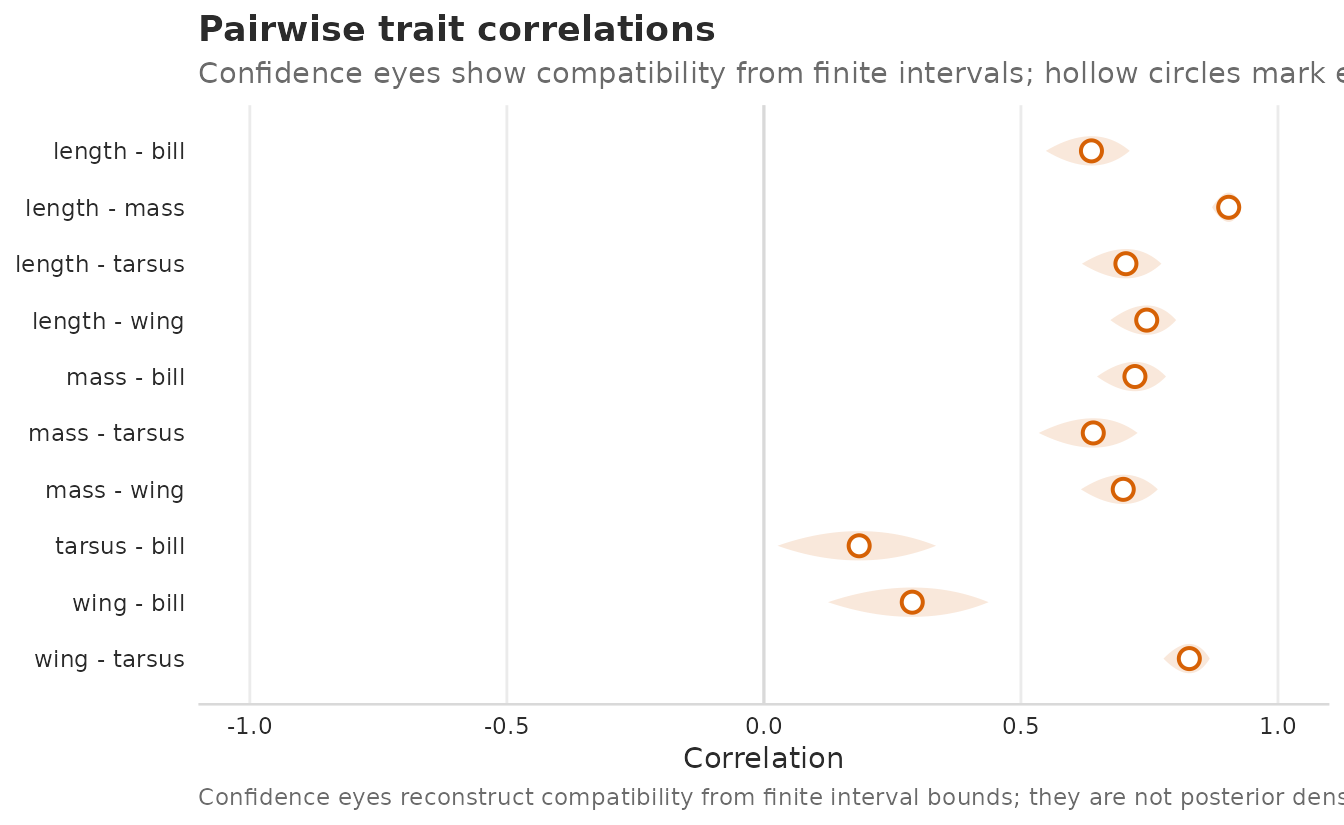
Pairwise between-individual trait correlations from a cached morphometrics bootstrap fixture. Confidence eyes reconstruct frequentist compatibility from percentile intervals; hollow circles mark estimates. Use a larger bootstrap run before making a final interval-calibration claim.
In this teaching fit, all unit-tier trait correlations are positive, so the figure mainly ranks their strength along the shared size axis. Tight confidence eyes near 1 are supplied by the cached bootstrap object (n_boot = 100, failed refits = 4). Treat this as a rendered plotting example; final inference should use a bootstrap size chosen for the study rather than the article’s lightweight fixture.
The same cached intervals can be shown as a matrix-style ellipse plot. Here the shape and fill show the sign and strength of each correlation; black borders and stars mark pairs whose supplied bootstrap interval does not cross zero. The stars are a display convention on the cached fixture’s percentile intervals, not a calibrated significance test — choose a bootstrap size for the study before drawing inferential conclusions.
plot(fit, type = "correlation_ellipse", level = "unit", boot = morph_boot_R)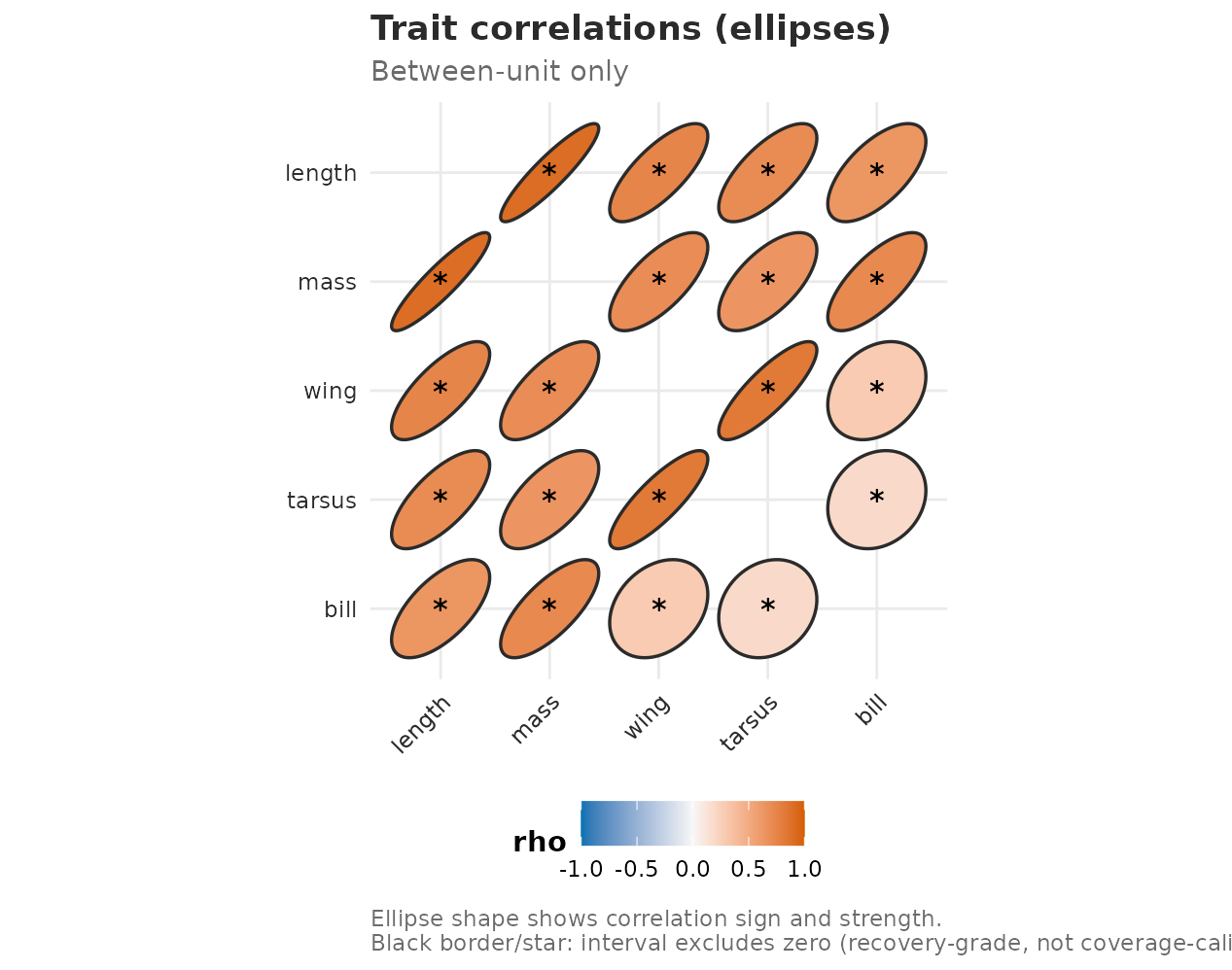
Ellipse view of the same cached between-individual correlation intervals. Ellipse tilt and colour show the fitted correlation; black borders and stars mark bootstrap percentile intervals that do not cross zero.
extract_communality(fit, level = "unit")
#> length mass wing tarsus bill
#> 0.8911057 0.9348032 0.8055346 0.8684255 0.7801652Communality is the fraction of trait ’s variance shared with the others via the latent factors; high means strongly integrated, low means trait-specific variation dominates.
This single generated data set is a teaching example, not a full simulation study. For this known fixture, the long and wide formulas recover the same off-diagonal covariance pattern. Broader coverage and interval-calibration claims require separate simulation studies across prespecified generating values, with every failed fit included in the accounting.
Why default latent()? dep and
indep on the same data
The morphometric workflow uses ordinary latent(d = K) to
estimate
.
Two alternative covstruct modes — dep (full unstructured
with
free entries) and indep (diagonal-only
,
no cross-trait modelling) — are also valid keywords. Refit the same data
under each to see what the user actually loses or gains. These are
ordinary unit-tier keywords; source-specific forms such as
phylo_dep() and spatial_dep() affect
phylogenetic or spatial unit-tier covariance instead.
fit_full_long <- gllvmTMB(
value ~ 0 + trait +
latent(0 + trait | individual, d = T),
data = df, trait = "trait", unit = "individual"
)
fit_full_wide <- gllvmTMB(
traits(length, mass, wing, tarsus, bill) ~ 1 +
latent(1 | individual, d = T),
data = df_wide, unit = "individual"
)
fit_dep_long <- gllvmTMB(
value ~ 0 + trait +
dep(0 + trait | individual),
data = df, trait = "trait", unit = "individual"
)
fit_dep_wide <- gllvmTMB(
traits(length, mass, wing, tarsus, bill) ~ 1 +
dep(1 | individual),
data = df_wide, unit = "individual"
)
fit_indep_long <- gllvmTMB(
value ~ 0 + trait +
indep(0 + trait | individual),
data = df, trait = "trait", unit = "individual"
)
fit_indep_wide <- gllvmTMB(
traits(length, mass, wing, tarsus, bill) ~ 1 +
indep(1 | individual),
data = df_wide, unit = "individual"
)
fit_full <- fit_full_long
fit_dep <- fit_dep_long
fit_indep <- fit_indep_long
cat("Max long/wide logLik difference across comparison fits:",
signif(max(abs(c(
as.numeric(logLik(fit_full_long)) - as.numeric(logLik(fit_full_wide)),
as.numeric(logLik(fit_dep_long)) - as.numeric(logLik(fit_dep_wide)),
as.numeric(logLik(fit_indep_long)) - as.numeric(logLik(fit_indep_wide))
))), 3),
"\n")
#> Max long/wide logLik difference across comparison fits: 0
S1 <- extract_Sigma(fit, level = "unit")$Sigma
S2 <- extract_Sigma(fit_full, level = "unit")$Sigma
S3 <- extract_Sigma(fit_dep, level = "unit")$Sigma
S4 <- extract_Sigma(fit_indep, level = "unit")$Sigma
off <- function(M) M[upper.tri(M)]
truth_off <- off(Sigma_true)
ll <- function(f) round(as.numeric(logLik(f)), 2)
data.frame(
mode = c("latent(d=2)", "latent(d=T)",
"dep", "indep"),
logL = c(ll(fit), ll(fit_full), ll(fit_dep), ll(fit_indep)),
off_diag_corr_truth = c(round(cor(off(S1), truth_off), 3),
round(cor(off(S2), truth_off), 3),
round(cor(off(S3), truth_off), 3),
NA),
diag_min_max = c(sprintf("%.2f - %.2f", min(diag(S1)), max(diag(S1))),
sprintf("%.2f - %.2f", min(diag(S2)), max(diag(S2))),
sprintf("%.2f - %.2f", min(diag(S3)), max(diag(S3))),
sprintf("%.2f - %.2f", min(diag(S4)), max(diag(S4))))
)
#> mode logL off_diag_corr_truth diag_min_max
#> 1 latent(d=2) -739.61 0.989 0.93 - 1.33
#> 2 latent(d=T) -739.45 0.989 0.93 - 1.33
#> 3 dep -739.45 0.989 0.87 - 1.26
#> 4 indep -1103.01 NA 0.93 - 1.33What this shows:
-
depandlatent(d = T)both recover the saturated covariance surface — the comparison is a useful check that the dense fit and the full-rank latent parameterisation agree on the covariance pattern.depandlatent(d = T)give the same saturated fit here — numerically equal logL and on this fixture. -
Reduced-rank
latent(d = 2)with default matches the saturated fit within 0.1 nats of logL. With traits and a rank-2 truth, the rank constraint loses essentially no information and gives the same off-diagonal correlation pattern as the saturated fit ( vs truth ≈ 0.95 for both). -
indeprecovers the diagonals fine — Gaussian gives per-trait identifiability for the marginal variance even with no cross-trait modelling — but loses every off-diagonal. The off-diagonal correlation with truth isNAbecause the fitted off-diagonals are exactly zero by construction. logL is materially worse.
For this simulated rank-2 Gaussian truth, default
latent(d = 2) and the saturated dep fit
recover nearly the same covariance pattern, while indep
cannot recover off-diagonal association because it sets those entries to
zero by construction. Pick latent(d = K) when you have a
strong
rank-
hypothesis (size + shape, here); fall back to dep if you
suspect
and want the saturated fit; reach for indep only when you
genuinely believe traits are mutually independent.
What the latent factors mean
Every factor model has a rotational ambiguity: and produce the same for any orthogonal . To compare loadings between fits or between packages, rotate to a standard convention (varimax for interpretability, or lower-triangular for a unique fit):
L_hat <- extract_ordination(fit, level = "unit")$loadings
rot <- rotate_loadings(
fit,
level = "unit",
method = "varimax",
anchor_traits = c("mass", "wing")
)
round(rot$Lambda, 2)
#> LV1 LV2
#> length 0.72 0.73
#> mass 0.67 0.89
#> wing 0.92 0.28
#> tarsus 0.92 0.14
#> bill 0.07 0.85The same convention is available as tidy rows when you want a table beside the figure:
loading_table <- extract_rotated_loadings_table(
fit,
level = "unit",
method = "varimax",
anchor_traits = c("mass", "wing"),
loading_scale = "standardized"
)
loading_table <- loading_table[
order(loading_table$axis, -loading_table$abs_loading),
c("trait", "axis", "loading", "axis_share", "anchor_trait")
]
head(loading_table, 8)
#> trait axis loading axis_share anchor_trait
#> 4 tarsus LV1 0.92167242 0.5537047 mass
#> 3 wing LV1 0.85907137 0.5537047 mass
#> 1 length LV1 0.66380729 0.5537047 mass
#> 2 mass LV1 0.57959588 0.5537047 mass
#> 5 bill LV1 0.06957319 0.5537047 mass
#> 10 bill LV2 0.88052530 0.4462953 wing
#> 7 mass LV2 0.77386811 0.4462953 wing
#> 6 length LV2 0.67116734 0.4462953 wing
ord <- extract_ordination(fit, level = "unit")
head(ord$scores)
#> LV1 LV2
#> 1 -0.4370449 1.29664647
#> 2 0.2975838 0.29257732
#> 3 2.4105980 -1.02649382
#> 4 2.0998041 -0.02503290
#> 5 1.3894629 0.10884484
#> 6 -0.4262655 0.03733953
plot(
fit,
type = "ordination",
level = "unit",
anchor_traits = c("mass", "wing"),
standardize_loadings = TRUE
) +
ggplot2::labs(
caption = paste(
"Grey points are latent scores; arrows are standardized loadings.",
"Axes use varimax rotation with sign anchors on mass and wing.",
"Use Sigma and correlation summaries for rotation-invariant interpretation.",
sep = "\n"
)
)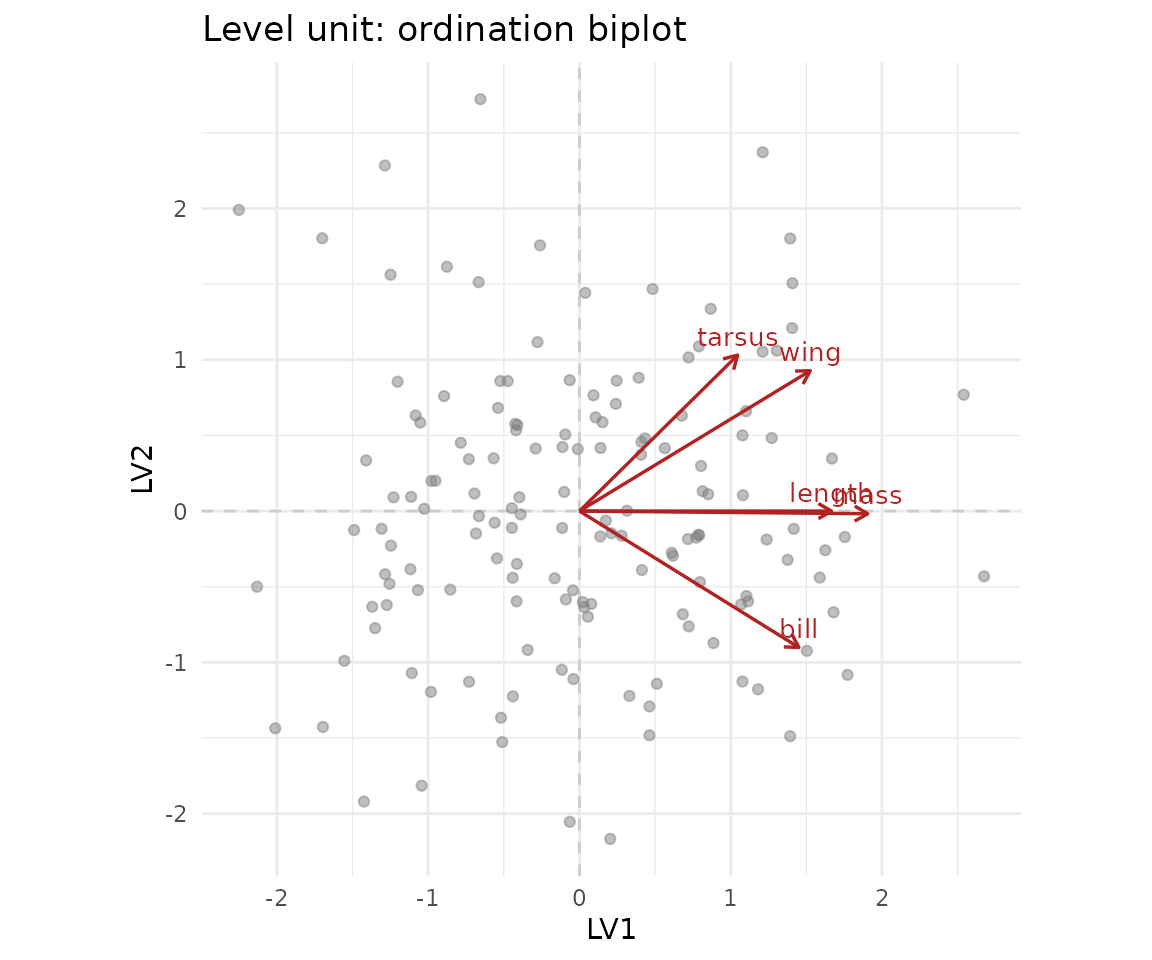
This ordination comes from the fitted covariance model (): unlike PCA or NMDS, the axes are estimated latent quantities rather than an eigen- or distance-decomposition of the raw data. Their uncertainty is not shown in this plot, and the grey points are predicted latent scores (empirical-Bayes conditional modes of ), not observed coordinates.
The biplot shows the trait loadings on the two latent axes. In a real
morphometric study, the first axis usually picks up overall
size, with all traits loading positively — the famous “general
factor” of body size. The second axis picks up shape contrasts. The sign
anchor is a reporting convention: here positive LV1 is anchored to
mass and positive LV2 to wing; changing signs
would not change the fitted covariance.
What this article does not cover
This is the simplest case on the complexity ladder. Richer models —
binary / joint species distribution
responses, phylogenetic and spatial dependence between rows, and
two-level between/within designs — build on this same
latent() foundation and are covered in their own guides as
they become public.
See also
- Covariance and correlation — why diagonal matters for interpreting trait correlations.
- Formula keyword grid — the syntax map for moving from this example to other covariance rows and modes.