Covariance and correlation: the model behind Sigma
Source:vignettes/articles/covariance-correlation.Rmd
covariance-correlation.RmdThis page is for the reader who has fitted a multivariate model and now asks: which traits vary together, and are the reported correlations too large if the model leaves out trait-specific variance?
The covariance, correlation, and communality extractors described here are part of the package’s experimental surface, and their interval machinery is still maturing.
Start from the model, not from the covariance matrix. For a Gaussian
stacked-trait GLLVM, the teaching model for individual i
and trait t is
The model has three moving parts. mu_t is the trait
mean, u_i is the individual’s latent position on the shared
axes, and psi_tt is trait-specific variance left over after
those shared axes are fitted. Sigma is not the model
itself; it is the trait covariance implied by that model.
The shared axes are identified only up to an orthogonal rotation and sign flip, so the individual loadings are not uniquely defined. But — and therefore , the correlations, and communality — are rotation-invariant, and are the quantities to report.
In ordinary latent() fits, gllvmTMB estimates both the
shared loading part and the diagonal Psi part by default. Use
latent(..., unique = FALSE) only when you deliberately want
the older loadings-only subset for a sensitivity check or compatibility
comparison.
This article is about Psi in a decomposed
latent() model. It is not a recommendation to use a
standalone diagonal term as the first model. When the goal is a
standalone marginal-only diagonal covariance, new examples should use
indep() so the intent is visible in the formula.
fit_no_residual <- gllvmTMB(
value ~ 0 + trait +
latent(0 + trait | individual, d = 2, unique = FALSE),
data = df, trait = "trait", unit = "individual"
)
fit_latent_default <- gllvmTMB(
value ~ 0 + trait + latent(0 + trait | individual, d = 2),
data = df, trait = "trait", unit = "individual"
)If your data are wide, with one row per individual and one column per
behaviour, use the same gllvmTMB() entry point with
traits(...) on the left-hand side:
fit_latent_default <- gllvmTMB(
traits(boldness, exploration, activity, aggression, sociability) ~
1 + latent(1 | individual, d = 2),
data = df_wide, unit = "individual"
)From model to Sigma
The fitted model above implies a covariance matrix for any covariance
tier you extract. For the ordinary latent() model, the
implied trait covariance is
Here level names the covariance tier being extracted:
unit for between-individual covariance,
unit_obs for within-individual or observation-level
covariance, and other tier names for structured terms.
is the
loading matrix from the latent() term.
is the
diagonal matrix of trait-specific variances.
The correlation matrix is a standardised version of
Sigma:
Both pieces matter. The diagonal of is — the sum of squared loadings plus the trait-specific diagonal variance — while the off-diagonals depend only on . The correlation divides those off-diagonals by the diagonal scale, so whether is present in the diagonal sets the scale of every reported correlation.
Read the model beside the R syntax and the report-ready summary:
| Object | R syntax or extractor | What the reader should report |
|---|---|---|
| Gaussian model | value ~ 0 + trait + latent(0 + trait | individual, d = 2) |
A reduced-rank multivariate model for the observed traits. |
level |
level = "unit" or level = "unit_obs"
|
Which covariance tier the summary belongs to. |
latent(..., d = K) |
Shared axes: traits that rise and fall together across units. | |
extract_Sigma_table(fit, level = "unit", part = "shared") |
Shared covariance explained by the latent variables only. | |
| and | default latent() Psi;
extract_Sigma_table(fit, level = "unit", part = "unique")
|
Trait-specific variance that keeps correlation denominators honest. |
extract_Sigma_table(fit, level = "unit", part = "total") |
Full trait covariance: shared structure plus trait-specific variance. | |
extract_correlations(fit, tier = "unit") or
$R from extract_Sigma()
|
The correlation matrix after standardising full . |
A side-by-side demonstration
We use a prepared Gaussian behavioural-syndrome example object. The
generator is
data-raw/examples/make-covariance-edge-cases-example.R; the
article uses the shipped object so the first thing the reader sees is
the model, not a long data-generating block.
The truth has both a shared low-rank component and a trait-specific diagonal Psi component. Fit the same data two ways:
-
Model A:latent(0 + trait | individual, d = 2, unique = FALSE)(no Psi) -
Model B:latent(0 + trait | individual, d = 2)(default Psi)
covex <- readRDS(system.file(
"extdata", "examples", "covariance-edge-cases-example.rds",
package = "gllvmTMB"
))
df <- covex$data_long
df_wide <- covex$data_wide
truth <- covex$truth
covex$story$question
#> [1] "Do five behaviours share latent syndrome axes while keeping behaviour-specific variance in the correlation denominator?"
head(df, 6)
#> individual trait value
#> 1 1 boldness 0.12828192
#> 2 1 exploration -0.03210224
#> 3 1 activity -0.56732317
#> 4 1 aggression -0.65759306
#> 5 1 sociability -0.35474482
#> 6 2 boldness -0.93719645The long-format formulas are:
covex$edge_cases$latent_only$formula_long
#> value ~ 0 + trait + latent(0 + trait | individual, d = 2, residual = FALSE)
# Model A is loadings-only (Psi switched off); Model B is the ordinary default
# (latent() carries the per-trait Psi). Written explicitly so the contrast is
# unambiguous under the 0.2.0 grammar where latent() carries Psi by default.
form_A <- value ~ 0 + trait + latent(0 + trait | individual, d = 2, unique = FALSE)
form_B <- value ~ 0 + trait + latent(0 + trait | individual, d = 2)
form_A
#> value ~ 0 + trait + latent(0 + trait | individual, d = 2, unique = FALSE)
form_B
#> value ~ 0 + trait + latent(0 + trait | individual, d = 2)The wide data-frame formula expresses the same recommended model
through the traits(...) left-hand side:
form_B_wide <- traits(boldness, exploration, activity, aggression, sociability) ~
1 + latent(1 | individual, d = 2)
form_B_wide
#> traits(boldness, exploration, activity, aggression, sociability) ~
#> 1 + latent(1 | individual, d = 2)
ctl <- gllvmTMBcontrol(se = FALSE)
fit_A <- gllvmTMB(
form_A,
data = df,
trait = covex$fit_args$trait,
unit = covex$fit_args$unit,
family = covex$fit_args$family,
control = ctl
)
fit_B <- gllvmTMB(
form_B,
data = df,
trait = covex$fit_args$trait,
unit = covex$fit_args$unit,
family = covex$fit_args$family,
control = ctl
)
fit_B_wide <- gllvmTMB(
form_B_wide,
data = df_wide,
unit = covex$fit_args$unit,
family = covex$fit_args$family,
control = ctl
)Long and wide fits use different data shapes, but here they represent the same likelihood:
c(
long = as.numeric(logLik(fit_B)),
wide = as.numeric(logLik(fit_B_wide))
)
#> long wide
#> -1111.151 -1111.151Now extract the implied and between-individual correlation matrix from each fit:
ext_A <- suppressMessages(
extract_Sigma(fit_A, level = "unit", part = "total")
)
ext_B <- extract_Sigma(fit_B, level = "unit", part = "total")Notice that the call on fit_A emits a one-shot
advisory explaining that unique = FALSE
returns
without Psi (we suppressed the message above for clean output but the
message is also stored in ext_A$note).
ext_A$note # advisory: diagonal Psi omitted by unique = FALSE
#> [1] "Sigma_unit is latent-only (Lambda Lambda^T): this fit used `latent(..., unique = FALSE)`, so trait-specific residual variance Psi is not modelled and correlations from this matrix overstate cross-trait coupling. For the full decomposition Sigma = Lambda Lambda^T + Psi, refit without `unique = FALSE` (the default)."The correlations side by side
cmp_A <- compare_Sigma_table(
fit_A,
truth = Sigma_true,
level = "unit",
measure = "correlation",
entries = "upper"
)
cmp_A$comparison <- "Model A: no-residual latent"
cmp_B <- compare_Sigma_table(
fit_B,
truth = Sigma_true,
level = "unit",
measure = "correlation",
entries = "upper"
)
cmp_B$comparison <- "Model B: ordinary latent with Psi"
corr_comparison <- rbind(cmp_A, cmp_B)
plot_Sigma_comparison(
corr_comparison,
measure = "correlation",
facet = "comparison",
sort = "trait"
)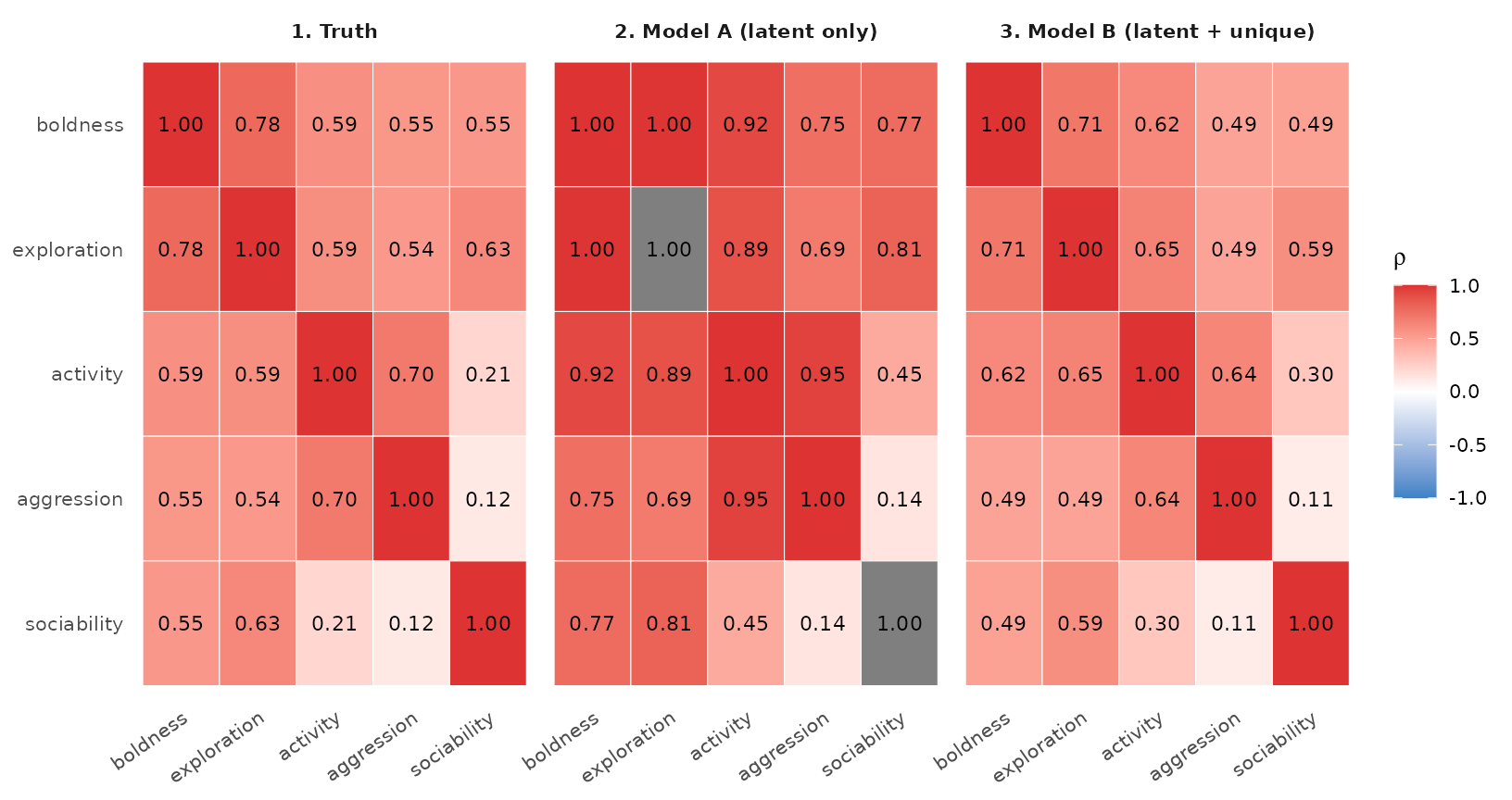
Trait-pair correlation errors for the no-residual latent subset and the ordinary latent model with Psi. Positive values mean the fitted correlation is larger than truth; zero means exact recovery for that pair.
Model A shows correlations that are uniformly
larger than truth. Model B pulls those errors back
toward zero. The mechanism: Model A’s
has small diagonals (only
),
so when we divide by
the off-diagonals get inflated. Model B’s
has the full
— diagonals at the right scale and correlations at the right
magnitude.
Report-ready Sigma rows
For reports, make the row object first. Each covariance target is one tidy row with trait names, matrix part, estimate, and interval placeholders:
sigma_rows_B <- extract_Sigma_table(
fit_B,
level = "unit",
part = "total",
measure = "covariance",
entries = "upper"
)
sigma_rows_B
#> estimand trait_i trait_j i j level
#> 1 Sigma_unit[boldness,exploration] boldness exploration 1 2 unit
#> 2 Sigma_unit[boldness,activity] boldness activity 1 3 unit
#> 3 Sigma_unit[exploration,activity] exploration activity 2 3 unit
#> 4 Sigma_unit[boldness,aggression] boldness aggression 1 4 unit
#> 5 Sigma_unit[exploration,aggression] exploration aggression 2 4 unit
#> 6 Sigma_unit[activity,aggression] activity aggression 3 4 unit
#> 7 Sigma_unit[boldness,sociability] boldness sociability 1 5 unit
#> 8 Sigma_unit[exploration,sociability] exploration sociability 2 5 unit
#> 9 Sigma_unit[activity,sociability] activity sociability 3 5 unit
#> 10 Sigma_unit[aggression,sociability] aggression sociability 4 5 unit
#> component matrix estimate lower upper interval_method interval_status
#> 1 total Sigma 1.07402353 NA NA none none
#> 2 total Sigma 0.81822969 NA NA none none
#> 3 total Sigma 0.82114973 NA NA none none
#> 4 total Sigma 0.62593991 NA NA none none
#> 5 total Sigma 0.61861566 NA NA none none
#> 6 total Sigma 0.92599601 NA NA none none
#> 7 total Sigma 0.70478522 NA NA none none
#> 8 total Sigma 0.80148129 NA NA none none
#> 9 total Sigma 0.20836227 NA NA none none
#> 10 total Sigma 0.09644977 NA NA none none
#> scale diagonal triangle
#> 1 latent FALSE upper
#> 2 latent FALSE upper
#> 3 latent FALSE upper
#> 4 latent FALSE upper
#> 5 latent FALSE upper
#> 6 latent FALSE upper
#> 7 latent FALSE upper
#> 8 latent FALSE upper
#> 9 latent FALSE upper
#> 10 latent FALSE upperpart = "total" is the default and is what you almost
always want for reporting. "shared" is useful if you
specifically want the latent-implied component (e.g. for ordination
interpretation or communality). "unique" gives the diagonal
of
as a named numeric vector.
The same table helper exposes the three decomposition parts without hand-indexing matrices:
sigma_part_rows_B <- rbind(
extract_Sigma_table(fit_B, level = "unit", part = "shared", entries = "diag"),
extract_Sigma_table(fit_B, level = "unit", part = "unique", entries = "diag"),
extract_Sigma_table(fit_B, level = "unit", part = "total", entries = "diag")
)
sigma_part_rows_B[c("component", "matrix", "trait_i", "trait_j", "estimate")]
#> component matrix trait_i trait_j estimate
#> 1 shared Sigma boldness boldness 1.0021803
#> 2 shared Sigma exploration exploration 1.1580281
#> 3 shared Sigma activity activity 1.1111363
#> 4 shared Sigma aggression aggression 0.7795385
#> 5 shared Sigma sociability sociability 0.7997145
#> 6 unique Psi boldness boldness 0.2852581
#> 7 unique Psi exploration exploration 0.2089832
#> 8 unique Psi activity activity 0.3743577
#> 9 unique Psi aggression aggression 0.2588508
#> 10 unique Psi sociability sociability 0.4533548
#> 11 total Sigma boldness boldness 1.2874383
#> 12 total Sigma exploration exploration 1.3670113
#> 13 total Sigma activity activity 1.4854940
#> 14 total Sigma aggression aggression 1.0383892
#> 15 total Sigma sociability sociability 1.2530693If you need a matrix for algebra checks, use
extract_Sigma() directly:
# Lambda Lambda^T alone (the "shared" component)
extract_Sigma(fit_B, level = "unit", part = "shared")$Sigma |> round(2)
#> boldness exploration activity aggression sociability
#> boldness 1.00 1.07 0.82 0.63 0.70
#> exploration 1.07 1.16 0.82 0.62 0.80
#> activity 0.82 0.82 1.11 0.93 0.21
#> aggression 0.63 0.62 0.93 0.78 0.10
#> sociability 0.70 0.80 0.21 0.10 0.80
# psi -- trait-specific diagonal variances (the `part = "unique"` component)
extract_Sigma(fit_B, level = "unit", part = "unique")$s |> round(2)
#> boldness exploration activity aggression sociability
#> 0.29 0.21 0.37 0.26 0.45
# Sigma_unit = Lambda Lambda^T + Psi (the "total" -- what you usually want)
extract_Sigma(fit_B, level = "unit", part = "total")$Sigma |> round(2)
#> boldness exploration activity aggression sociability
#> boldness 1.29 1.07 0.82 0.63 0.70
#> exploration 1.07 1.37 0.82 0.62 0.80
#> activity 0.82 0.82 1.49 0.93 0.21
#> aggression 0.63 0.62 0.93 1.04 0.10
#> sociability 0.70 0.80 0.21 0.10 1.25
plot_Sigma_table(sigma_rows_B, sort = "magnitude")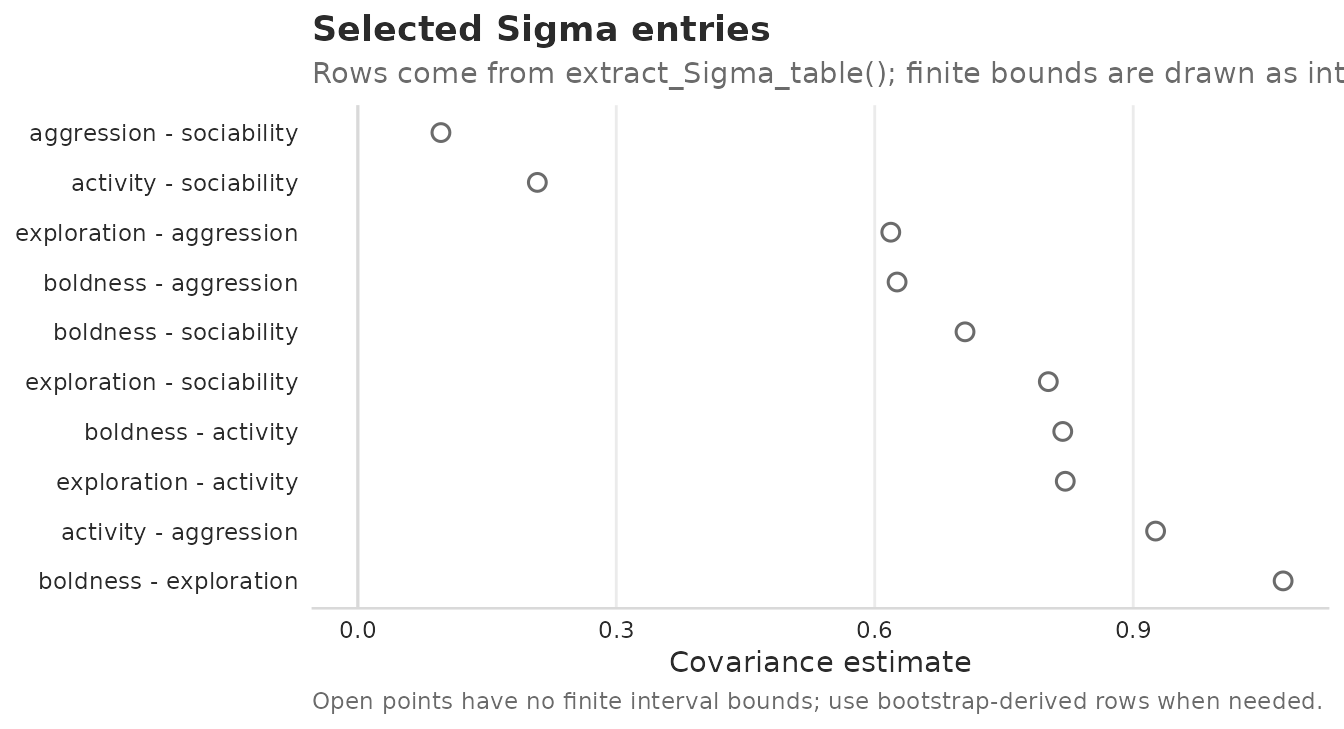
Upper-triangle off-diagonal entries of Sigma_unit from the ordinary latent model with Psi. Open points mark point estimates without finite interval bounds; this figure does not add uncertainty beyond the rows supplied to the plot.
Communality and ICC need the full too
Two trait-level summaries used in behavioural-syndromes and phenotypic-integration workflows both depend on the full decomposition. If you compute them from a no-residual latent fit you get the wrong answer.
Communality
This is the fraction of trait ’s variance explained by the shared latent variables. It is bounded between 0 and 1 and is a proportion-of-variance summary, analogous in scale to (heritability) or . Like the other -derived summaries it is a model-based factor-analytic quantity of the fitted GLLVM — computed from the fitted covariance decomposition, not a PCA or exploratory-factor-analysis eigen-decomposition of the raw data.
For a reader-facing table, the alignment is:
| Symbol | R output | Interpretation |
|---|---|---|
extract_communality(fit, level = "unit") |
Fraction of trait t variance explained by the shared
latent variables. |
|
| numerator |
extract_Sigma(fit, level = "unit", part = "shared")
diagonal |
Shared variance for trait t. |
| denominator |
extract_Sigma(fit, level = "unit", part = "total")
diagonal |
Shared variance plus trait-specific diagonal variance. |
The catch: with no-residual latent fits,
for every trait by construction, so
for every trait — the communality is identically 1 and tells you
nothing. Ordinary latent() gives the denominator a
per-trait
slot by default, and communality becomes informative.
extract_communality(fit_A, level = "unit") # no-residual fit: all = 1
#> boldness exploration activity aggression sociability
#> 1 1 1 1 1
extract_communality(fit_B, level = "unit") # ordinary latent with Psi
#> boldness exploration activity aggression sociability
#> 0.7784297 0.8471240 0.7479911 0.7507189 0.6382045For the report surface, keep the exact table but display the rows as
a matrix. extract_correlations() supplies the point
estimates and Fisher-z interval bounds; plot_correlations()
arranges those supplied values for reading, with estimates in the upper
triangle and interval bounds in the lower triangle:
corr_B <- extract_correlations(fit_B, tier = "unit")
corr_B
#> tier trait_i trait_j correlation lower upper method interval_status
#> 1 B boldness exploration 0.80958888 NA NA none none
#> 2 B boldness activity 0.59166593 NA NA none none
#> 3 B exploration activity 0.57623662 NA NA none none
#> 4 B boldness aggression 0.54136395 NA NA none none
#> 5 B exploration aggression 0.51922402 NA NA none none
#> 6 B activity aggression 0.74557884 NA NA none none
#> 7 B boldness sociability 0.55488888 NA NA none none
#> 8 B exploration sociability 0.61237831 NA NA none none
#> 9 B activity sociability 0.15272006 NA NA none none
#> 10 B aggression sociability 0.08455389 NA NA none none
plot_correlations(
corr_B,
style = "heatmap",
matrix_layout = "estimate_ci",
title = "Ordinary latent trait correlations",
caption = "Fill shows estimates; lower labels show supplied Fisher-z bounds."
)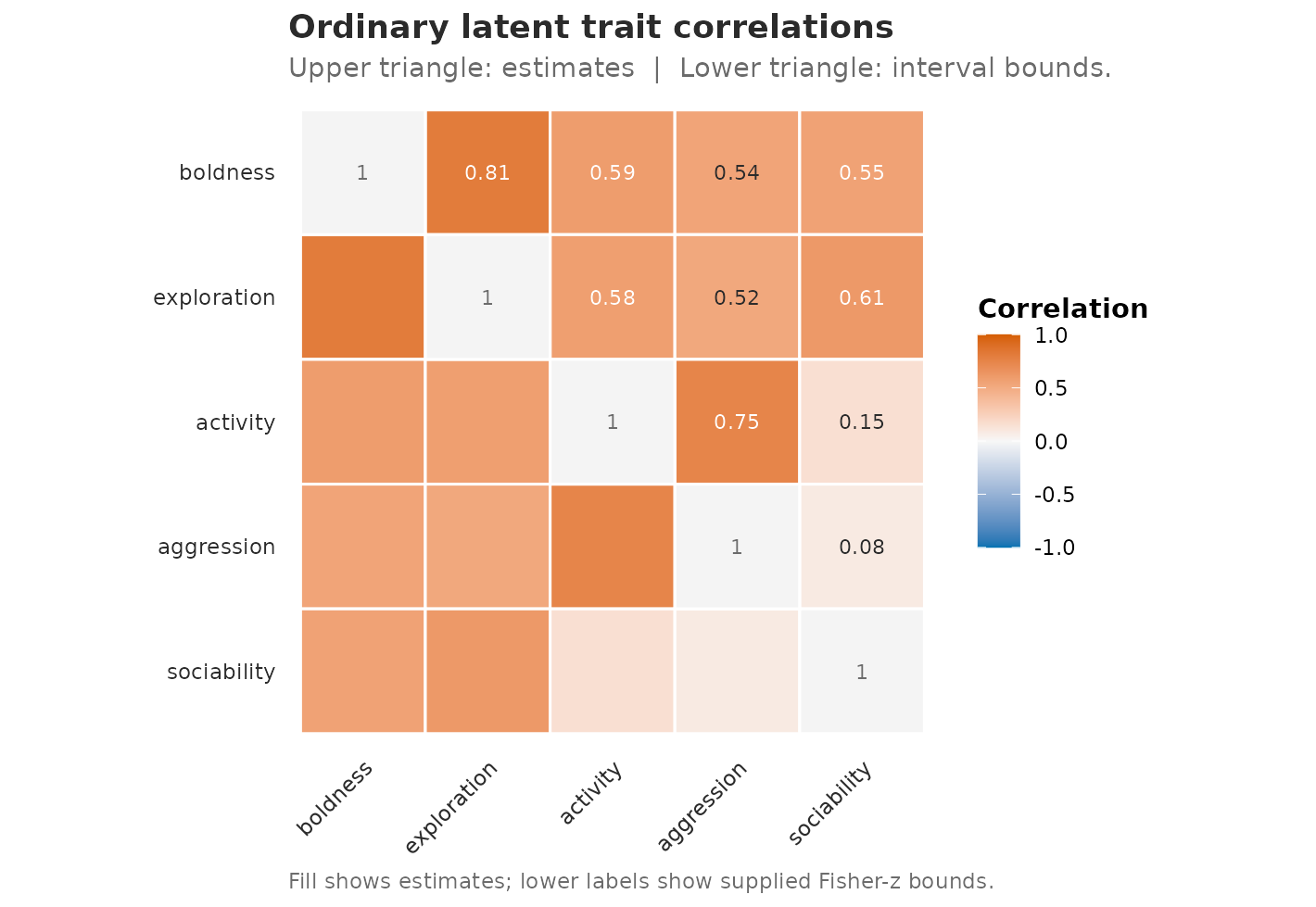
Pairwise correlations from the ordinary latent model with Psi. Upper cells show point estimates; lower cells display the Fisher-z interval columns already present in the extractor output.
Read this as a formatted table, not as a new uncertainty calculation.
It is a display of the rows returned by
extract_correlations(): it does not bootstrap, profile, or
calibrate uncertainty — it only renders the Fisher-z interval columns
already present in corr_B. Those bounds are themselves
nominal Fisher-z
(Wald-on-)
intervals: their empirical coverage has not been calibrated and they do
not establish repeated-sampling coverage for gllvmTMB’s latent
covariances, so read them as an uncertainty display, not a calibrated
interval.
Site-level / individual-level ICC
For two-level fits the site-level (individual-level) ICC is
i.e. the proportion of total trait variance attributable to between-
unit differences. Each piece needs the full decomposition for its level
—
and
.
Ordinary latent() supplies that diagonal Psi at each level
by default, so each level needs its own latent() term for
to be honest.
In gllvmTMB this is (pattern, not run in this article)
extract_ICC_site(fit)— see the recommended two-level
latent() pattern below.
Binomial responses: the link’s implicit residual
Binary outcomes (family = binomial()) are a special
case. The link function fixes a latent-scale residual variance:
for logit,
for probit,
for cloglog. This implicit residual already plays the role
of
on the latent scale, so an explicit indep() diagonal term
on top of a binary fit is typically not identified.
extract_Sigma() has a
link_residual = "auto" option that adds the link-specific
implicit residual to
for the marginal latent-scale interpretation:
# (illustrative — not run; needs a binary fit)
extract_Sigma(fit_binary, level = "unit", part = "total",
link_residual = "auto")For continuous traits, the recommended starting pattern is ordinary
latent(), which includes the diagonal Psi companion by
default. For count, mixed-family, phylogenetic, and spatial fits, use
the same idea only after checking that the fit converged; see the
corresponding family article for worked examples.
Two-level (between + within) models: two latent()
terms
For repeated-measures data the recommended pattern is one ordinary
latent() term at each covariance level (Nakagawa &
Schielzeth 2010; Nakagawa, Johnson & Schielzeth 2017):
fit_two_level <- gllvmTMB(
value ~ 0 + trait +
latent(0 + trait | individual, d = d_B) +
latent(0 + trait | obs_id, d = d_W),
data = df,
trait = "trait",
unit = "individual",
unit_obs = "obs_id"
)giving (behavioural syndromes — between-individual covariance) and (integrated plasticity — within-individual covariance). Each level has its own default diagonal Psi.
Observation-level random effects for non-Gaussian fits
Overdispersed Poisson is the one family that carries
both an estimated observation-level random effect (OLRE) and a
link residual, so it is the case where adding a latent-scale diagonal
component makes sense (Nakagawa & Schielzeth 2010). For
nbinom2, tweedie, and the other overdispersed
count families the overdispersion is already baked into the single
distribution-specific residual
— adding an OLRE on top would double-count it. The OLRE pattern is
described here but not run in this article:
The
on the latent log scale plays the role of
.
Native OLRE support is available: add an obs_id column (one
level per row), include indep(0 + trait | obs_id) in the
formula, pass unit_obs = "obs_id", and use
extract_residual_split(fit) to separate
(the estimated OLRE variance) from
(the distribution-specific latent residual; Nakagawa & Schielzeth
2010; Nakagawa, Johnson & Schielzeth 2017).
Summary
| You want… | You need | Notes |
|---|---|---|
| Cross-trait correlations on a Gaussian / lognormal / Gamma fit | ordinary latent() at every level |
latent(..., unique = FALSE) omits Psi and can inflate
correlations. |
| Correlations on a binary fit |
latent() only; link_residual = "auto" for
marginal scale |
Implicit residual depends on link (π²/3, 1, π²/6). |
| Phylogenetic decomposition | folded phylo_latent(..., unique = TRUE) for the
phylogenetic tier; ordinary latent() for the
non-phylogenetic species tier |
,
with each
using shared + Psi pieces. |
| Communality |
extract_communality(fit) + the right Psi-aware
pattern |
Communality formula uses Σ; needs full Σ to be meaningful. |
Lambda Lambda^T only |
extract_Sigma(fit, part = "shared") |
Useful for ordination. |
Psi / psi only |
extract_Sigma(fit, part = "unique") |
Trait-specific diagonal variances as a vector. |
See also
-
Cross-family trait
correlations, including nominal responses — one shared latent factor
across Gaussian, binary, count, ordinal, and nominal traits, and the
reference-invariant summary for a
multinomial()trait - Pitfalls — common mistakes including correlation inflation
-
?latent— the ordinary Psi-carrying latent keyword -
?extract_Sigma— the unified extractor -
?suggest_lambda_constraint— a helper for confirmatory loading structures - Formula keyword grid — syntax map for moving from this decomposition to other covariance rows
- Nakagawa & Schielzeth (2010) Repeatability for Gaussian and non-Gaussian data: a practical guide for biologists. Biological Reviews 85(4):935–956. https://doi.org/10.1111/j.1469-185X.2010.00141.x
- Westneat, Wright & Dingemanse (2015) The biology hidden inside residual within-individual phenotypic variation. Biological Reviews 90, 729–743. https://doi.org/10.1111/brv.12131