Joint species distribution models for binary occurrence data
Source:vignettes/articles/joint-sdm.Rmd
joint-sdm.RmdAfter an environmental gradient is in the model, which species still tend to occur together, and which species avoid each other? That is the first joint species distribution model (JSDM) question this page answers.
The data are binary occurrence records: one row per site, one column
or trait per species, and a 0/1 response for absence/presence. The model
uses latent() to estimate a low-rank residual co-occurrence
structure across species after env_1 is accounted for.
This example supports both long data and the wide
traits(...) formula, using a binomial-logit model with
latent(0 + trait | site, d = K) for residual co-occurrence.
The Fisher-z intervals below are model-based uncertainty diagnostics;
their repeated-sampling coverage has not been established for this model
class. Single-species spatial SDMs belong in sdmTMB, and
multivariate spatial fields via spatial_*() are beyond the
scope of this introductory JSDM example.
Load and fit
The article uses a shipped teaching fixture rather than building the
data inside the vignette. It contains a complete
Site x Species occurrence matrix: 120 sites, 8 species, 2
latent residual co-occurrence axes, and an environmental covariate
env_1 with species-specific responses. The true loadings
put the species into four niche quadrants (dry-low, dry-high, wet-low,
wet-high), with two species per quadrant.
jsdm_path <- system.file(
"extdata", "examples", "joint-sdm-example.rds",
package = "gllvmTMB"
)
source_candidates <- file.path(
c(".", "..", "../.."),
"inst", "extdata", "examples", "joint-sdm-example.rds"
)
input_file <- knitr::current_input(dir = TRUE)
if (!is.null(input_file)) {
source_candidates <- c(
source_candidates,
file.path(
dirname(dirname(dirname(input_file))),
"inst", "extdata", "examples", "joint-sdm-example.rds"
)
)
}
candidate_paths <- c(jsdm_path, source_candidates)
candidate_paths <- candidate_paths[nzchar(candidate_paths)]
jsdm_path <- candidate_paths[file.exists(candidate_paths)][1]
if (is.na(jsdm_path)) {
stop("Could not find joint-sdm-example.rds.")
}
jsdm <- readRDS(jsdm_path)
df <- jsdm$data_long
df_wide <- jsdm$data_wideThe same JSDM fits in both long format and the
traits(...) wide format. The long form is shown first; the
wide form is the formula-API equivalent on a pivoted data frame. Both
reach the same engine.
# Long format -- one row per (site, species) observation
fit_jsdm <- gllvmTMB(
value ~ 0 + trait + (0 + trait):env_1 +
latent(0 + trait | site, d = 2),
data = df,
trait = "trait",
unit = "site",
family = binomial()
)
fit_jsdm
#> Stacked-trait gllvmTMB fit
#> Traits = 8, site = 120
#> Covstructs: latent_unit, indep_unit
#> Fixed effects (b_fix): 16
#> ML log L = 11765.408 convergence = 0
#> Note: Lambda_B identified up to rotation (use suggest_lambda_constraint() or rotate_loadings()).
#> Run gllvmTMB_diagnose(fit) for a full health check, or summary(fit) for parameter estimates.
# Wide format -- one row per site, one column per species. The
# `traits(...)` LHS names the species columns; the compact RHS
# shorthand uses `1 + env_1`, which expands internally to
# `0 + trait + (0 + trait):env_1`.
fit_jsdm_wide <- gllvmTMB(
traits(sp_1, sp_2, sp_3, sp_4, sp_5, sp_6, sp_7, sp_8) ~
1 + env_1 + latent(1 | site, d = 2),
data = df_wide,
unit = "site",
family = binomial()
)
all.equal(logLik(fit_jsdm), logLik(fit_jsdm_wide), tolerance = 1e-8)
#> [1] TRUELong or wide data shape
JSDM is the package’s natural long-format use case because each row
already carries (site, species) and the response factor
trait indexes species. When the data already live as one
row per site and one binary column per species, the
traits(...) wide form is the same model through the same
gllvmTMB() entry point. In either shape, true absences
should be explicit zeros. Missing response cells are not a safe shortcut
for absences, because missingness can also mean the site or species was
not surveyed.
What latent() estimates
For binary occurrence data with a logit link, the fitted linear predictor is
The site score is the unobserved residual state of site , and the species loading says how species responds to those residual axes. The rotation of is arbitrary, so the stable co-occurrence target is the species covariance implied by the loadings. For this single-trial Bernoulli fixture, each species-site cell contributes one binary observation. The default between-site diagonal companion is therefore not separately identified from the link scale and the engine maps it off:
For a binary logit JSDM, correlations are easier to interpret on the
latent-liability scale.
extract_correlations(link_residual = "auto") adds the fixed
logistic residual variance
to the diagonal:
The logistic residual term is fixed by the binomial logit link, not
estimated from the data. Ordinary latent() normally carries
a diagonal
companion, but the fit message explains why that default component is
mapped off for these single-trial cells. Multi-trial binomial data or
genuine repeated observations can identify additional variation; they
are different designs and should not be inferred from this occurrence
matrix.
Where loading constraints fit
The loading matrix is usually exploratory in a JSDM: its rotation is arbitrary, while is the interpretable co-occurrence target. If you have an a-priori hypothesis that a species group defines an axis, or that a species should have zero loading on an axis, that is a confirmatory loading question rather than the default species-distribution workflow.
gllvmTMB supports that confirmatory path with
lambda_constraint = list(unit = M), where NA
means “estimate this loading” and a number means “pin this loading
during fitting.” Numeric pins are exact model restrictions: because the
latent scores have unit variance, a loading pinned to 1 is
assumed to equal one; it is not a cosmetic sign convention. The current
route acts on the packed lower-triangular loading matrix, so it is not a
general free-form confirmatory-factor interface.
Specify a biological loading hypothesis before inspecting this fitted loading matrix, verify that the matrix rows follow the response order, and stop if the constrained fit is unhealthy. A matrix suggested from the same data can provide a reproducible descriptive orientation, but it remains exploratory and does not turn selected zeros into biological facts. This guide therefore keeps correlations and as its interpretation targets.
Latent-scale residual species correlations
Pairwise species correlations on the latent-liability scale. The
opt-in method = "fisher-z" gives Fisher-transform heuristic
bounds that are fast (seconds) and bounded inside
by construction. method = "wald" is an accepted alias for
the same Fisher-z path. Two heavier alternatives are also available:
method = "bootstrap" re-fits the model under a parametric
bootstrap for a full sampling-distribution CI, and
method = "profile" inverts the profile likelihood
pair-by-pair (cost scales as
constrained refits).
One scale caveat is worth stating up front, because it trips people
up on binary fits. The default link_residual = "auto" puts
the correlation on the latent-liability scale — it adds
the fixed logistic residual
to the diagonal of
before forming the correlation, which is the interpretable co-occurrence
quantity for a JSDM. fisher-z, wald, and
bootstrap all honour that argument. The current
profile path, however, always reports the correlation of
the bare
(it ignores link_residual), so its point estimate is the
undiluted latent-factor correlation, not the latent-liability one. The
optional comparison snippets below make the difference explicit for
readers who want to audit the scale.
What is and is not certified: the point estimates and Fisher-z bounds are useful diagnostics for this fitted example, but the package-wide calibrated-coverage work is still open. Treat intervals here as model-based uncertainty summaries, not as a claim that every binary or mixed-family covariance interval has reached nominal coverage.
corr_rows <- extract_correlations(
fit_jsdm,
tier = "unit",
method = "fisher-z",
link_residual = "auto"
)
corr_rows
#> tier trait_i trait_j correlation lower upper method
#> 1 B sp_1 sp_2 0.153555913 -0.02641239 0.32388284 fisher-z
#> 2 B sp_1 sp_3 0.128632141 -0.05180359 0.30093506 fisher-z
#> 3 B sp_2 sp_3 0.018398708 -0.16137481 0.19699041 fisher-z
#> 4 B sp_1 sp_4 -0.038128913 -0.21589477 0.14208346 fisher-z
#> 5 B sp_2 sp_4 -0.093762798 -0.26849182 0.08693966 fisher-z
#> 6 B sp_3 sp_4 0.153546600 -0.02642192 0.32387430 fisher-z
#> 7 B sp_1 sp_5 -0.028984725 -0.20714987 0.15104131 fisher-z
#> 8 B sp_2 sp_5 0.072350398 -0.10829533 0.24837082 fisher-z
#> 9 B sp_3 sp_5 -0.288700390 -0.44491851 -0.11543233 fisher-z
#> 10 B sp_4 sp_5 -0.197555473 -0.36391070 -0.01898645 fisher-z
#> 11 B sp_1 sp_6 -0.115408285 -0.28867805 0.06518139 fisher-z
#> 12 B sp_2 sp_6 -0.044896452 -0.22234849 0.13543477 fisher-z
#> 13 B sp_3 sp_6 -0.209613817 -0.37477434 -0.03155816 fisher-z
#> 14 B sp_4 sp_6 -0.077285072 -0.25302139 0.10338847 fisher-z
#> 15 B sp_5 sp_6 0.168001108 -0.01158921 0.33709169 fisher-z
#> 16 B sp_1 sp_7 -0.140516818 -0.31190245 0.03972505 fisher-z
#> 17 B sp_2 sp_7 -0.164262535 -0.33367947 0.01543320 fisher-z
#> 18 B sp_3 sp_7 0.054151117 -0.12631626 0.23114890 fisher-z
#> 19 B sp_4 sp_7 0.139373438 -0.04088938 0.31084930 fisher-z
#> 20 B sp_5 sp_7 -0.146835893 -0.31771526 0.03328138 fisher-z
#> 21 B sp_6 sp_7 0.001381071 -0.17790431 0.18057771 fisher-z
#> 22 B sp_1 sp_8 -0.202881734 -0.36871483 -0.02453251 fisher-z
#> 23 B sp_2 sp_8 -0.201163228 -0.36716576 -0.02274189 fisher-z
#> 24 B sp_3 sp_8 -0.023443364 -0.20183658 0.15645540 fisher-z
#> 25 B sp_4 sp_8 0.124534776 -0.05595559 0.29714335 fisher-z
#> 26 B sp_5 sp_8 -0.096574066 -0.27112226 0.08412346 fisher-z
#> 27 B sp_6 sp_8 0.058834221 -0.12169041 0.23559113 fisher-z
#> 28 B sp_7 sp_8 0.217567191 0.03988110 0.38191498 fisher-z
#> interval_status
#> 1 heuristic_unvalidated
#> 2 heuristic_unvalidated
#> 3 heuristic_unvalidated
#> 4 heuristic_unvalidated
#> 5 heuristic_unvalidated
#> 6 heuristic_unvalidated
#> 7 heuristic_unvalidated
#> 8 heuristic_unvalidated
#> 9 heuristic_unvalidated
#> 10 heuristic_unvalidated
#> 11 heuristic_unvalidated
#> 12 heuristic_unvalidated
#> 13 heuristic_unvalidated
#> 14 heuristic_unvalidated
#> 15 heuristic_unvalidated
#> 16 heuristic_unvalidated
#> 17 heuristic_unvalidated
#> 18 heuristic_unvalidated
#> 19 heuristic_unvalidated
#> 20 heuristic_unvalidated
#> 21 heuristic_unvalidated
#> 22 heuristic_unvalidated
#> 23 heuristic_unvalidated
#> 24 heuristic_unvalidated
#> 25 heuristic_unvalidated
#> 26 heuristic_unvalidated
#> 27 heuristic_unvalidated
#> 28 heuristic_unvalidatedThe matrix view below shows the same point estimates as a
species-by-species map. For a binary JSDM, the useful first picture is
the latent-liability correlation pattern after adding
the fixed logistic link residual to the denominator; raw
entries can be dominated by loading scale and are harder to interpret.
The Fisher-z interval bounds stay in corr_rows rather than
being printed inside every matrix cell.
plot_correlations(
corr_rows,
style = "heatmap",
matrix_layout = "by_level",
label_type = "estimate",
label_digits = 2,
include_diagonal = TRUE,
title = "Residual species correlations",
subtitle = "Latent-liability correlations after accounting for env_1."
) +
ggplot2::labs(caption = NULL)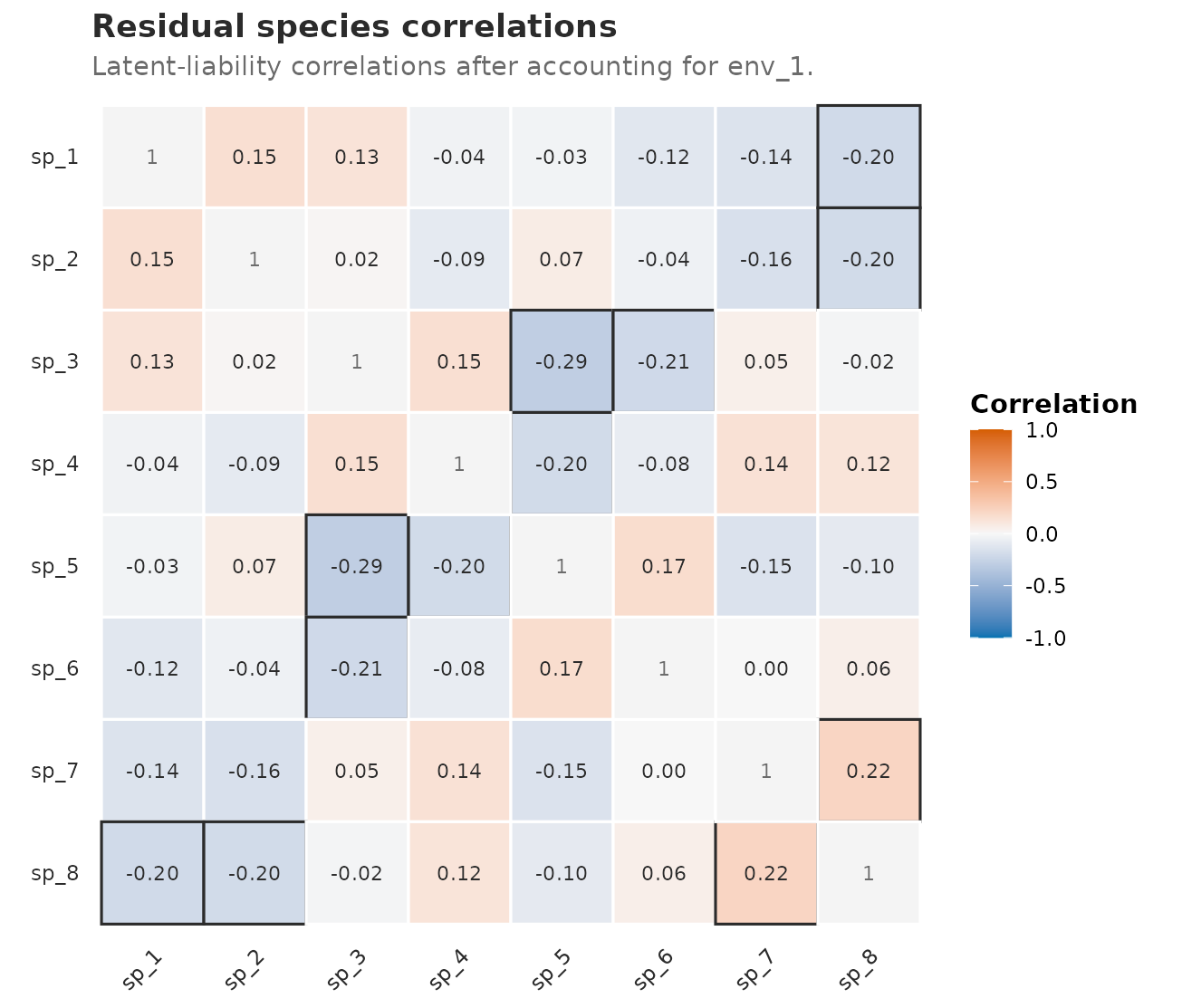
Residual species correlations from the fitted binary JSDM. Cells show Fisher-z point estimates on the latent-liability scale after accounting for env_1 and the fixed logistic link residual; interval bounds are available in the preceding table.
Use these correlations, rather than the signs of individual loadings, for the rotation-invariant co-occurrence story.
Confidence intervals on the binary latent scale
This section works through the three CI methods on two estimands — a pairwise species correlation and a per-species ICC — to show what each method returns and where it currently stops short. These chunks are optional because the bootstrap and profile paths re-fit the model; run them when the interval method itself is the question, not for a first pass through the article.
Three CI methods for one correlation
Take a single species pair (sp_1, sp_2) and
ask for the same correlation three ways. Keeping
link_residual = "auto" for the Fisher-z and bootstrap calls
holds them on the latent-liability scale, so their point estimates agree
and only the interval differs:
pair_ij <- c("sp_1", "sp_2")
cor_fisher <- extract_correlations(
fit_jsdm, tier = "unit", pair = pair_ij,
method = "fisher-z", link_residual = "auto"
)
cor_boot <- extract_correlations(
fit_jsdm, tier = "unit", pair = pair_ij,
method = "bootstrap", nsim = 20, seed = 1, link_residual = "auto"
)
rbind(cor_fisher, cor_boot)[, c("trait_i", "trait_j",
"correlation", "lower", "upper", "method")]The Fisher-z and bootstrap point estimates match because both add the logistic residual to the diagonal; the bootstrap interval is the empirical 2.5/97.5 percentile across the refits.
The profile method is the one that reports a
different number, because it currently works on the bare
scale. To compare it fairly, request the Fisher-z interval with
link_residual = "none" so both sit on the same (undiluted)
scale:
cor_profile <- extract_correlations(
fit_jsdm, tier = "unit", pair = pair_ij,
method = "profile"
)
cor_fisher_bare <- extract_correlations(
fit_jsdm, tier = "unit", pair = pair_ij,
method = "fisher-z", link_residual = "none"
)
rbind(cor_profile, cor_fisher_bare)[, c("trait_i", "trait_j",
"correlation", "lower", "upper", "method")]Now the two point estimates agree: the profile correlation matches
the bare-scale Fisher-z value. The profile interval is a
likelihood-ratio inversion around that point — but note it can come back
as NA when the constrained refit fails to bracket the bound
(a near-boundary correlation here is a common cause), which is the same
not-yet-calibrated CI story flagged above. The take-home is not
that one method is wrong, but that profile and the
default latent-liability correlations live on different scales
until the profile path learns to honour link_residual. For
the interpretable JSDM co-occurrence story, prefer the latent-liability
scale (auto), and use Fisher-z for speed or the bootstrap
when you want an empirical sampling-distribution interval.
ICC (communality) on the latent scale
For a single-trial binary JSDM, the natural communality quantity is the share of each species’ latent-liability variance represented by the shared factors rather than the fixed logistic residual:
The unit-level latent() term estimates the shared
loading covariance, and link_residual = "auto" adds the
fixed logistic residual variance
,
so
extract_communality(level = "unit", link_residual = "auto")
returns exactly this ratio — the proportion of the latent-liability
variance that the shared factors explain. (This is not the
between-vs-within extract_repeatability() ICC, which needs
paired between- and within-unit diagonal tiers such as
indep(0 + trait | site) + indep(0 + trait | site_species);
a single-trial occurrence matrix has no within-site replication for
that, so extract_repeatability() and
extract_ICC_site() are not applicable to this fit.)
icc_point <- extract_communality(
fit_jsdm, level = "unit", link_residual = "auto"
)
round(icc_point, 3)
#> sp_1 sp_2 sp_3 sp_4 sp_5 sp_6 sp_7 sp_8
#> 0.193 0.152 0.323 0.143 0.310 0.143 0.195 0.266A parametric bootstrap puts an interval on each species’ ICC:
icc_boot <- extract_communality(
fit_jsdm, level = "unit", link_residual = "auto",
ci = TRUE, method = "bootstrap", nsim = 20, seed = 1
)
icc_boot[, c("trait", "c2", "lower", "upper", "method")]The intervals are wide — eight species and 120 sites is modest information for a per-species variance share, and the upper bounds for the more strongly-loading species run close to 1.
Two limitations apply to the ICC interval in this example:
-
Wald is not implemented for communality (it is a
non-linear function of several parameters); requesting
method = "wald"emits a note and falls back to the bootstrap. -
Profile bounds are not yet delivered for this
proportion on a single-trial binary fit — the constrained refit fails to
bracket and the interval returns
NA. The point estimate is still the correct latent-scale ICC; only the profile interval is missing.
The point estimates are available, but interval coverage on binary and mixed-family scales has not been established. Report the point estimate and the interval method used; do not present these intervals as coverage-certified.
Ordination biplot
Site scores on the two latent variables (LV1, LV2), with species
loadings shown as arrows. This is model-based ordination: LV1
and LV2 are the latent variables of a fitted GLLVM, estimated jointly
with the env_1 regression — not a variance-maximising PCA,
a distance-based NMDS embedding, or a covariate-constrained CCA/RDA of
the raw occurrence matrix. The plotted site scores are predicted
(empirical-Bayes) conditional modes of the residual site states
:
shrunk toward zero, and carrying prediction uncertainty the point
display does not show. The rotation-invariant interpretation target is
the model-implied residual co-occurrence
(defined above), not the axis coordinates themselves — species loading
in the same direction tend to co-occur after env_1 is
accounted for.
plot(
fit_jsdm,
type = "ordination",
level = "unit",
rotation = "varimax",
sign_anchor = "auto",
standardize_loadings = TRUE
) +
ggplot2::labs(
title = "Residual species ordination",
subtitle = "Varimax-rotated site scores and species loading directions.",
caption = NULL
)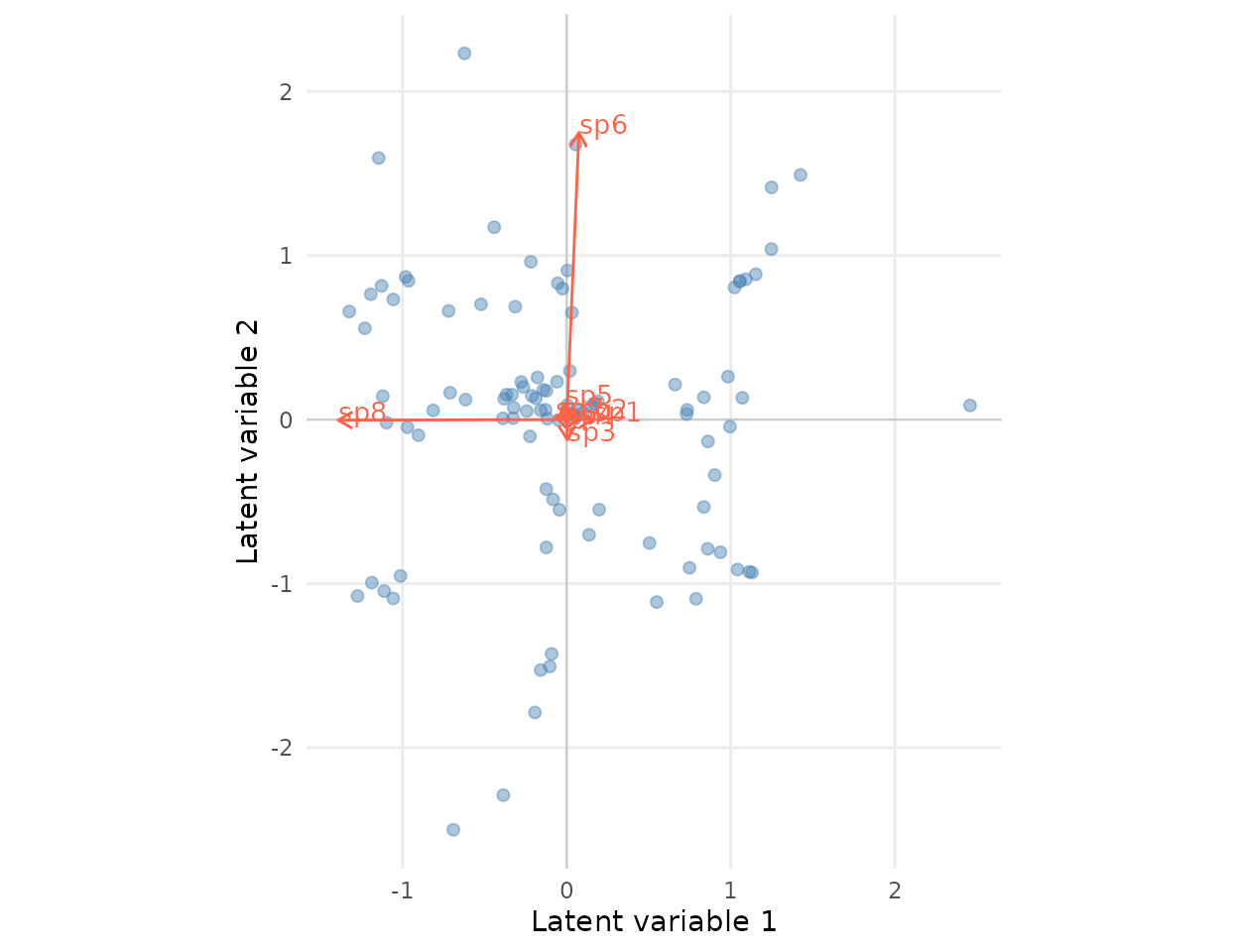
Ordination biplot from the fitted binary JSDM, using varimax-rotated and sign-anchored site scores and species loadings. Loading orientation and sign are not identifiable; the model-implied Sigma and correlations remain the rotation-invariant interpretation target.
The fixture’s truth places the eight species into four
designed association groups: sp_1 /
sp_2, sp_3 / sp_4,
sp_5 / sp_6, and sp_7 /
sp_8. The fitted arrows should be read for broad grouping,
not axis labels. The exact orientation is arbitrary — only
is identifiable. Species arrows pointing in the same direction co-occur
after env_1 is accounted for; arrows pointing in opposite
directions tend to mutually exclude.
See also
-
Morphometrics — Gaussian
stacked-trait analogue with ordinary
latent(). -
Choosing latent rank
— how to compare candidate values of
d. - Loading-constraint helpers —
?confirmatory_lambdaand?suggest_lambda_constraintdocument the advanced API contracts. -
vignette("response-families")— full table of supported families and the per-family .
References
- Niku, J., Warton, D.I., Hui, F.K.C., & Taskinen, S. (2017). Generalized linear latent variable models for multivariate count and biomass data in ecology. Journal of Agricultural, Biological, and Environmental Statistics 22, 498–522. https://link.springer.com/article/10.1007/s13253-017-0304-7
- Niku, J., Hui, F.K.C., Taskinen, S., & Warton, D.I. (2019). gllvm: Fast analysis of multivariate abundance data with generalized linear latent variable models in r. Methods in Ecology and Evolution 10, 2173–2182. https://doi.org/10.1111/2041-210X.13303
- Nakagawa, S., & Schielzeth, H. (2010). Repeatability for Gaussian and non-Gaussian data: a practical guide for biologists. Biological Reviews 85, 935–956. https://doi.org/10.1111/j.1469-185X.2010.00141.x
- Nakagawa, S., Johnson, P.C.D., & Schielzeth, H. (2017). The coefficient of determination R² and intra-class correlation coefficient from generalized linear mixed-effects models revisited and expanded. Journal of the Royal Society Interface 14,
- Warton, D.I., Blanchet, F.G., O’Hara, R.B., Ovaskainen, O., Taskinen, S., Walker, S.C., & Hui, F.K.C. (2015). So many variables: joint modeling in community ecology. Trends in Ecology & Evolution 30, 766–779. https://doi.org/10.1016/j.tree.2015.09.007