Functional biogeography GLLVM: M1 to M2 to extensions
Source:vignettes/articles/functional-biogeography.Rmd
functional-biogeography.RmdThe replicated site–species–trait GLLVM is the canonical functional-biogeography model of Nakagawa et al. (in prep). This article fits it as a two-step model ladder:
- §1 – M1 (functional integration core). Trait covariation decomposed into a between-site component (global functional integration) and a within-site component (local functional integration). No space, no phylogeny – just the syndrome story.
- §2 – M2 (controls for space and phylogeny). Re-fit M1 with spatial and phylogenetic adjustment terms. Compare M2’s / to M1’s. If the syndrome story holds, the controls have done their job.
- §3 – Extensions. Promote space or phylogeny to its own covariance decomposition (M3, M4) when the inferential target is spatial or phylogenetically conserved trait syndromes. We preview the syntax and point at companion articles; the fits themselves live elsewhere.
A family of models
Before §1, the conceptual scaffolding. The replicated site–species–trait GLLVM is one of a family of trait-covariance models. The right model depends on the response shape:
| Data / question | Recommended model |
|---|---|
| Species occurrences/abundances; traits explain species responses | Site
species JSDM / fourth-corner GLLVM (joint-sdm) |
| One CWM/functional summary per site and trait | Site-summary / CWM site trait GLLVM |
| Species-in-site trait observations, enough within-site richness | Replicated site–species–trait GLLVM (this article) |
| Species have one trait value each (AVONET-style) | Two-stage site-summary model (corvidae-two-stage) |
| Trait-state counts or functional-gene frequencies | Trait-state abundance/count GLLVM |
| Trait integration across species/evolution | Species
trait phylogenetic GLLVM (phylogenetic-gllvm) |
This article models direct trait observations on multiple species at each site. The other rows of the table point at the right article for the other response shapes; do not force this model onto the wrong one.
A second conceptual move runs through §1 and §2: the adjustment vs partitioning distinction for spatial and phylogenetic terms.
-
Adjustment models (M2, this article’s main focus).
Spatial and phylogenetic terms are controls, so the primary
biological targets remain
and
.
Use
spatial_unique()andphylo_unique()– per-trait variances on a shared correlation structure ( and ). -
Partitioning models (M3, M4 in §3). Spatial and
phylogenetic signals get their own reduced-rank covariance decomposition
(,
)
via
spatial_latent + spatial_uniqueandphylo_latent + phylo_unique. The question shifts to: are syndromes spatially structured or phylogenetically conserved? These models are more data-demanding; treat them as sensitivity analyses.
Setup and DGP
Wide-format input. The data here are naturally long (one row per
(site, species, trait)cell), but field datasets sometimes arrive wide (one row per(site, species), one column per trait). The formula-wide path keeps that shape readable:traits(trait_1, ..., trait_T) ~ 1 + latent(1 | site_species, d = 2) + unique(1 | site_species). The long form below stays canonical because it keepssite,species, andsite_speciesvisible for the crossed ecological interpretation.
A 60-site, 30-species, 6-trait fixture with mean 5 species per site (300 rows-ish) gives both M1 and M2 enough information to recover the simulation truth on the largest covariance entries. The DGP includes:
- a rank-2 between-site truth (): two trait syndromes with opposite signs across the two trait blocks (1-3 vs 4-6);
- a rank-1 within-site truth (): a single shared local axis;
- small phylogenetic and spatial signals so M2 has something to control for, but small enough that the M1-vs-M2 comparison is honest – the syndrome story should not be an artefact of geography or shared ancestry.
set.seed(42)
n_sites <- 60
n_species <- 30
n_traits <- 6
tree <- ape::rcoal(n_species)
tree$tip.label <- paste0("sp", seq_len(n_species))
## Two clear syndromes (rank d_B = 2 truth)
Lambda_B_true <- matrix(c(
0.9, 0.7, 0.6, -0.2, -0.5, -0.3, # axis 1 (traits 1-3 high; 4-6 low)
0.2, -0.1, 0.0, 0.7, 0.7, 0.5 # axis 2 (traits 4-6 high)
), nrow = n_traits, ncol = 2)
Lambda_W_true <- matrix(c(0.5, 0.4, 0.3, 0.4, 0.3, 0.2),
nrow = n_traits, ncol = 1)
S_B_true <- rep(0.15, n_traits)
S_W_true <- rep(0.20, n_traits)
sim <- simulate_site_trait(
n_sites = n_sites, n_species = n_species, n_traits = n_traits,
mean_species_per_site = 5,
Lambda_B = Lambda_B_true, Lambda_W = Lambda_W_true,
S_B = S_B_true, S_W = S_W_true,
Cphy = ape::vcv(tree, corr = TRUE),
sigma2_phy = rep(0.10, n_traits),
sigma2_sp = rep(0.05, n_traits),
spatial_range = 0.4,
sigma2_spa = rep(0.10, n_traits),
sigma2_eps = 0.10,
seed = 42
)
df <- sim$data
levels(df$species) <- tree$tip.label
Sigma_B_true <- Lambda_B_true %*% t(Lambda_B_true) + diag(S_B_true)
Sigma_W_true <- Lambda_W_true %*% t(Lambda_W_true) + diag(S_W_true)§1 – M1: functional integration core
The lead-in model. No space, no phylogeny. The mean structure is
a per-trait intercept
plus per-trait slopes
on two site-level predictors env_1, env_2. The
random-effect structure is
with
- , – global functional integration, between-site trait covariance.
- , – local functional integration, within-site species deviations.
- – residual.
gllvmTMB syntax:
t0 <- Sys.time()
fit_M1 <- gllvmTMB(
value ~ 0 + trait +
(0 + trait):env_1 + (0 + trait):env_2 +
latent(0 + trait | site, d = 2) +
unique(0 + trait | site) +
latent(0 + trait | site_species, d = 1) +
unique(0 + trait | site_species),
data = df,
cluster = "species"
)
t_M1 <- round(as.numeric(Sys.time() - t0, units = "secs"), 1)
fit_M1$opt$convergence
#> [1] 0Wall time: 2.9 s. Extract and :
Sigma_B_M1 <- extract_Sigma(fit_M1, level = "unit", part = "total")$Sigma
Sigma_W_M1 <- extract_Sigma(fit_M1, level = "unit_obs", part = "total")$Sigma
round(Sigma_B_M1, 2)
#> trait_1 trait_2 trait_3 trait_4 trait_5 trait_6
#> trait_1 1.01 0.68 0.57 -0.21 -0.47 -0.15
#> trait_2 0.68 0.82 0.47 -0.30 -0.52 -0.24
#> trait_3 0.57 0.47 0.57 -0.28 -0.46 -0.22
#> trait_4 -0.21 -0.30 -0.28 0.65 0.53 0.34
#> trait_5 -0.47 -0.52 -0.46 0.53 0.99 0.44
#> trait_6 -0.15 -0.24 -0.22 0.34 0.44 0.59
round(Sigma_W_M1, 2)
#> trait_1 trait_2 trait_3 trait_4 trait_5 trait_6
#> trait_1 0.73 0.22 0.15 0.21 0.17 0.15
#> trait_2 0.22 0.58 0.13 0.18 0.15 0.13
#> trait_3 0.15 0.13 0.44 0.13 0.11 0.09
#> trait_4 0.21 0.18 0.13 0.62 0.14 0.12
#> trait_5 0.17 0.15 0.11 0.14 0.47 0.10
#> trait_6 0.15 0.13 0.09 0.12 0.10 0.48The block structure is recovered: traits 1-3 covary positively, traits 4-6 covary positively, and the two blocks covary negatively – matching the rank-2 simulation truth. Per-trait communality and the site-level ICC :
round(extract_communality(fit_M1, level = "unit"), 3)
#> trait_1 trait_2 trait_3 trait_4 trait_5 trait_6
#> 0.948 0.672 0.712 0.626 0.756 0.493
round(extract_ICC_site(fit_M1), 3)
#> trait_1 trait_2 trait_3 trait_4 trait_5 trait_6
#> 0.582 0.583 0.566 0.512 0.677 0.549Three things to watch for in any M1 fit, each a separate identifiability concern:
Note (1) – vs identifiability. With no within-cell replication (one observation per (
site,species,trait) triple), only the sum is identified. The engine handles this by auto-suppressingsigma_epsto a tiny floor whenever a per-rowunique()is present (you’ll see a one-shot inform when M1 fits). Replication within cell – multiple individuals of the same species at the same site, or repeated visits – is the design move that splits them. Seepitfallsfor a worked demonstration.
Note (2) – communality is rank-conditional. depends on the choice of . Refitting M1 at on the same data gives different communalities than at :
fit_M1_d1 <- gllvmTMB(
value ~ 0 + trait +
(0 + trait):env_1 + (0 + trait):env_2 +
latent(0 + trait | site, d = 1) +
unique(0 + trait | site) +
latent(0 + trait | site_species, d = 1) +
unique(0 + trait | site_species),
data = df,
cluster = "species"
)
rbind(
d_B_1 = round(extract_communality(fit_M1_d1, level = "unit"), 3),
d_B_2 = round(extract_communality(fit_M1, level = "unit"), 3)
)
#> trait_1 trait_2 trait_3 trait_4 trait_5 trait_6
#> d_B_1 0.628 0.677 0.727 0.417 0.665 0.304
#> d_B_2 0.948 0.672 0.712 0.626 0.756 0.493The two communality vectors disagree by 0.05–0.4 across traits. The reason is structural: is the diagonal of over the diagonal of , and shrinking by one takes a column out of . Always report the rank you used. Pick on a model-comparison criterion (AIC, cross-validated likelihood) before reporting communality, not after.
Note (3) – the response shape determines the question. This article uses direct trait observations . For occurrence/abundance data with traits as covariates, fit a JSDM / fourth-corner GLLVM (
joint-sdm). For one CWM-style summary per site, use the site-summary CWM model. For AVONET-style data with one trait value per species, use the two-stage workflow. The replicated site–species–trait model below is right for our DGP; do not force it onto a different one.
§2 – M2: spatial and phylogenetic adjustment
Same equation as §1 but with four extra random-effect terms:
with
- – per-trait Matérn spatial field on the SPDE mesh; spatial control.
- – per-trait phylogenetic species random effect on the shared ; phylogenetic control.
- – per-trait non-phylogenetic species random effect (independent across species); the residual species effect after phylogeny.
These are the adjustment-mode terms: each trait carries its own variance on a shared correlation structure (, , ), without introducing a cross-trait spatial or phylogenetic loading matrix. This keeps the focal interpretation on and .
The
pair is the same Hadfield–Nakagawa (2010) decomposition used in the phylogenetic-gllvm
article, just placed at the cluster (species) slot of a crossed
(site
species) fit rather than at the unit slot of a per-species fit. The two
correlation matrices
and
are orthogonal, so
and
are jointly identifiable – see the note after the fit for the empirical
regime.
mesh <- make_mesh(df, c("lon", "lat"), cutoff = 0.1)
t0 <- Sys.time()
fit_M2 <- gllvmTMB(
value ~ 0 + trait +
(0 + trait):env_1 + (0 + trait):env_2 +
spatial_unique(0 + trait | coords, mesh = mesh) +
latent(0 + trait | site, d = 2) +
unique(0 + trait | site) +
latent(0 + trait | site_species, d = 1) +
unique(0 + trait | site_species) +
unique(0 + trait | species) + # q_it
phylo_unique(species, tree = tree), # p_it
data = df,
cluster = "species"
)
t_M2 <- round(as.numeric(Sys.time() - t0, units = "secs"), 1)
fit_M2$opt$convergence
#> [1] 0Wall time: 18.2 s.
A note on identifiability. With
and a crossed (site
species) design,
recovers within $$10% relative error and
within $$50% (per-trait estimates can be noisy when one trait’s
phylogenetic signal is weak; the trait-averaged
is more stable). At small
or weak phylogenetic signal, the
and
pieces cannot be cleanly separated and one should compare M1 (without
unique(0 + trait | species)) to M2 (with it) to confirm
both terms contribute on the focal data. The engine prints a one-shot
informational note pointing at this regime when it detects the
pairing.
The pedagogical moment: do the controls move the syndrome?
Re-extract and from M2 and compare to M1’s:
Sigma_B_M2 <- extract_Sigma(fit_M2, level = "unit", part = "total")$Sigma
Sigma_W_M2 <- extract_Sigma(fit_M2, level = "unit_obs", part = "total")$Sigma
R_B_M1 <- extract_Sigma(fit_M1, level = "unit")$R
R_B_M2 <- extract_Sigma(fit_M2, level = "unit")$R
R_W_M1 <- extract_Sigma(fit_M1, level = "unit_obs")$R
R_W_M2 <- extract_Sigma(fit_M2, level = "unit_obs")$R
c(max_abs_diff_R_B = round(max(abs(R_B_M2 - R_B_M1)), 3),
max_abs_diff_R_W = round(max(abs(R_W_M2 - R_W_M1)), 3))
#> max_abs_diff_R_B max_abs_diff_R_W
#> 0.157 0.166Side-by-side correlation-matrix heatmaps:
heatmap_df <- function(R, label) {
rn <- rownames(R) %||% paste0("trait_", seq_len(nrow(R)))
data.frame(
model = label,
trait_i = factor(rep(rn, ncol(R)), levels = rn),
trait_j = factor(rep(rn, each = nrow(R)), levels = rn),
rho = as.vector(R)
)
}
R_B_long <- rbind(
heatmap_df(R_B_M1, "M1 (no controls)"),
heatmap_df(R_B_M2, "M2 (+ space + phylo)")
)
ggplot(R_B_long, aes(trait_i, trait_j, fill = rho)) +
geom_tile(colour = "white") +
geom_text(aes(label = sprintf("%.2f", rho)), size = 2.8) +
scale_fill_gradient2(low = "#1f78b4", mid = "white", high = "#b2182b",
midpoint = 0, limits = c(-1, 1)) +
facet_wrap(~ model) +
coord_equal() +
labs(x = NULL, y = NULL,
title = expression("Between-site trait correlations " *
R[B] * " = cor(" * Sigma[B] * ")")) +
theme_minimal(base_size = 11) +
theme(panel.grid = element_blank(),
axis.text.x = element_text(angle = 45, hjust = 1))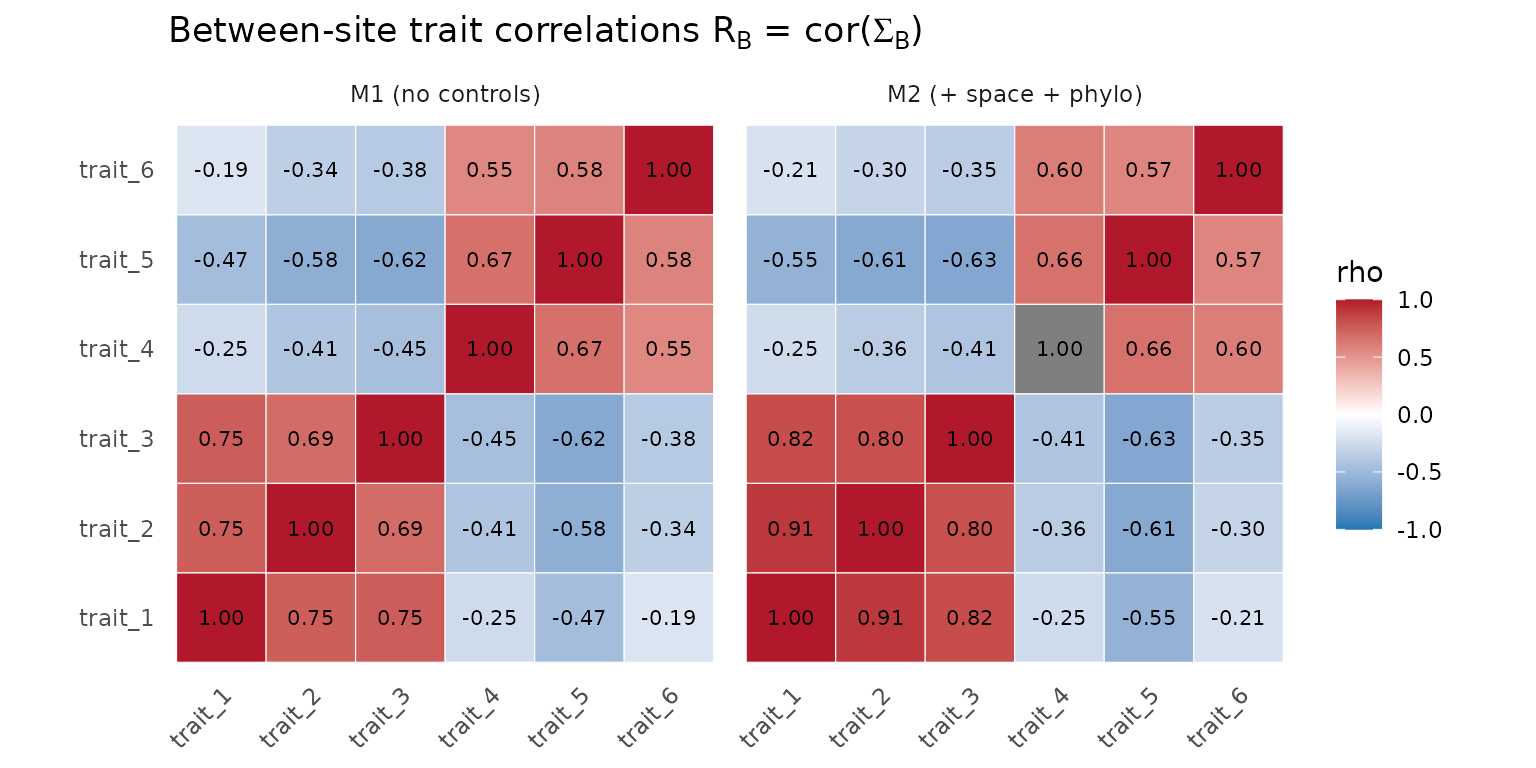
Cross-trait correlation matrix at the between-site (Sigma_B) tier under M1 (no controls) and M2 (spatial + phylogenetic adjustment). The block structure (traits 1-3 vs 4-6) is preserved across models.
R_W_long <- rbind(
heatmap_df(R_W_M1, "M1 (no controls)"),
heatmap_df(R_W_M2, "M2 (+ space + phylo)")
)
ggplot(R_W_long, aes(trait_i, trait_j, fill = rho)) +
geom_tile(colour = "white") +
geom_text(aes(label = sprintf("%.2f", rho)), size = 2.8) +
scale_fill_gradient2(low = "#1f78b4", mid = "white", high = "#b2182b",
midpoint = 0, limits = c(-1, 1)) +
facet_wrap(~ model) +
coord_equal() +
labs(x = NULL, y = NULL,
title = expression("Within-site trait correlations " *
R[W] * " = cor(" * Sigma[W] * ")")) +
theme_minimal(base_size = 11) +
theme(panel.grid = element_blank(),
axis.text.x = element_text(angle = 45, hjust = 1))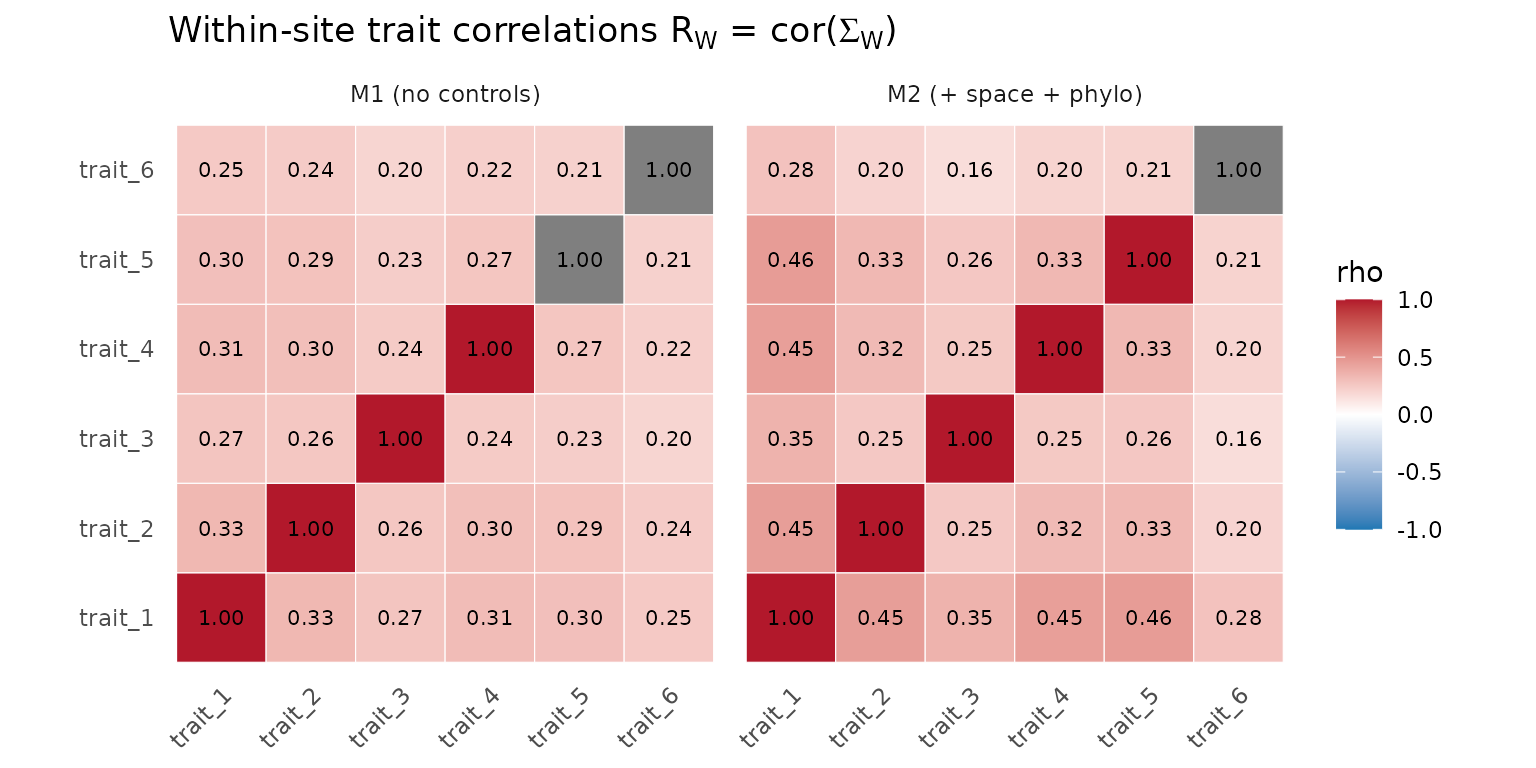
Cross-trait correlation matrix at the within-site (Sigma_W) tier under M1 and M2. The local-integration story is similarly stable to the addition of controls.
The block structure of – traits 1-3 positively correlated, traits 4-6 positively correlated, the two blocks negatively correlated – is the same under M1 and M2. That is the desired finding. It tells the reader the syndrome was not a geography or ancestry artefact: even after controlling for the spatial and phylogenetic structure that the data carry, the same two syndromes survive.
If the comparison instead showed M2’s flattening (syndrome erased) or shifting sign (syndrome reversed), you would have a different and equally interesting story: the apparent global functional integration was largely a function of spatial covariation in the environmental drivers, or of shared ancestry. Either finding should be reported – the M1 vs M2 comparison is the diagnostic for which.
Sample size guidance. M2 is a multi-tier model with five random-effect tiers and ~70 free parameters at . Identifiability requires enough groups at each tier, not just enough total observations:
- for stable
- mean species per site for identifiable (with single observation per (site, species, trait) cell, only is identifiable – see Note 1 in §1)
- for the per-trait phylogenetic () and non-phylogenetic species () variances
- phylogenetic signal at moderate-or-stronger () for the phylogenetic decomposition to bite
Below these thresholds, fall back to M1 – drop the controls, report and as partial (not “fully adjusted”) global and local integration. Don’t add tiers your data can’t support: gllvmTMB makes complex models expressible, but expressibility is not identifiability. A formal regime map across M1–M4 is in preparation (
dev/design/11-identifiability-regime-map.md); a dedicated article will follow once the simulation grid completes.
§3 – Extensions: when space and phylogeny become the target
When the question is about space or phylogeny itself – “are syndromes spatially structured?”, “is one of the syndromes phylogenetically conserved?” – promote the corresponding signal from a per-trait control to a full reduced-rank covariance decomposition. We sketch the syntax for two extension models below and point at the dedicated articles for the worked fits.
M3 – spatial trait syndromes
Add a spatial_latent term in addition to
spatial_unique to estimate
,
the spatial trait-covariance decomposition. Loadings on
tell you which traits hang together spatially.
fit_M3 <- gllvmTMB(
value ~ 0 + trait + (0 + trait):env_1 + (0 + trait):env_2 +
spatial_latent(0 + trait | coords, d = 2, mesh = mesh) +
spatial_unique(0 + trait | coords, mesh = mesh) +
latent(0 + trait | site, d = 2) +
unique(0 + trait | site) +
latent(0 + trait | site_species, d = 1) +
unique(0 + trait | site_species) +
phylo_unique(species, tree = tree),
data = df, cluster = "species"
)The spatial term is the same engine as the phylogenetic two-piece
decomposition documented in phylogenetic-gllvm. M3
needs more spatial signal than M2 to identify both
and
separately; treat its loadings as a sensitivity analysis to M2’s
per-trait spatial variances. A worked example will appear in the planned
spatial-trait-syndromes article.
M4 – phylogenetically conserved syndromes
Symmetric to M3, but on the phylogeny side: pair
phylo_latent with phylo_unique to estimate
,
the phylogenetic two-piece decomposition. Loadings on
tell you which traits cluster together along the tree.
fit_M4 <- gllvmTMB(
value ~ 0 + trait + (0 + trait):env_1 + (0 + trait):env_2 +
spatial_unique(0 + trait | coords, mesh = mesh) +
latent(0 + trait | site, d = 2) +
unique(0 + trait | site) +
latent(0 + trait | site_species, d = 1) +
unique(0 + trait | site_species) +
phylo_latent(species, d = 2, tree = tree) +
phylo_unique(species, tree = tree),
data = df, cluster = "species", unit = "species" # PGLLVM mode
)The two-piece phylogenetic fit is the focus of the phylogenetic-gllvm
article and its diagnostic counterpart two-U-phylogeny. That
article also covers the identifiability conditions:
(
in phylo_latent) is identifiable up to rotation, and the
two-piece split between
and
requires either
or a constraint matrix.
What we did not fit
We deliberately do not promote the ladder beyond M4 in this article. The “kitchen-sink” model that combines latent partitioning on every tier (, , , ) is conceptually beautiful but statistically fragile – the partitioning at one tier trades off against the others, and the loadings stop being separately identifiable without strong constraints. M2 is the sweet spot for publication-grade fits; M3 and M4 are sensitivity analyses; beyond that, the data have to carry the burden of identifiability that the model can no longer.
Diagnostics
sanity_multi(fit_M2)
#> Optimiser convergence (== 0): PASS
#> Max |gradient| < 1.0e-02: PASS (max |gr| = 0.00332)
#> Hessian positive-definite: PASS
#> Max fixed-effect SE < 100: WARN (max SE = 2.58e+04)
#> latent(site, d=B) diag loadings non-zero: PASS (min |Lambda_B diag| = 0.135)
#> latent(site_species, d=W) diag loadings non-zero: PASS (min |Lambda_W diag| = 0.61)Convergence, max gradient, Hessian PD-ness, fixed-effect SE, and
near-zero loading diagonals – the report flags any rank that was set
higher than the data support. The fixed-effect SE check is the loosest
filter: in fits with a phylogenetic random effect the SE column also
picks up some derived sd-report quantities and can flag WARN even when
the underlying fixed-effect estimates are well- behaved (compare to the
tidy(fit_M2, "fixed") output). The other four checks are
the ones to act on.
See also
-
phylogenetic-gllvm– the two-piece phylogenetic decomposition (M4 above) at species level. -
joint-sdm– non-Gaussian JSDM / fourth-corner model: traits explain species responses to environment. -
corvidae-two-stage– AVONET-style data where each species has one trait value. -
pitfalls– common gotchas: factor level order, rotation, the DGP-must-match-the-fit rule. -
profile-likelihood-ci– CIs on , entries via profile likelihood, Wald, or parametric bootstrap. -
simulation-recovery– multi-replicate recovery study for the full ladder.