How to choose your model: a decision tree
Source:vignettes/articles/choose-your-model.Rmd
choose-your-model.RmdgllvmTMB covers a large model space — one or two
observational levels, optional phylogenetic structure, optional spatial
structure, Gaussian / binary / Poisson / mixed responses, reduced-rank
or full-rank trait covariance. That generality is helpful when your data
are unusual but unhelpful when you are picking a formula on day 1.
This article is the decision tree we wish we had: five questions about your data, each answered with a small piece of the formula, that you assemble at the end into the model that matches your problem. If you already know the rung you want, jump to the linked article for that rung.
How to use this article
Walk down the five questions in order. At each step you pick one option, which gives you a piece of the formula or the call. By the end of section 5 you have all the pieces; section 6 shows the canonical assembled formulas for the most common combinations.
The five questions:
- What does each row represent?
- What is the response distribution?
- Are rows phylogenetically related?
- Are rows spatially organised?
- Do you want a reduced-rank story for trait covariance?
The complexity ladder, at a glance
Each rung adds one structural piece to the previous. Pick the highest
rung you actually need — extra structure costs identifiability and fit
time. The recovery study
(articles/simulation-recovery.html) quantifies how cleanly
each rung’s parameters are recovered at modest n.
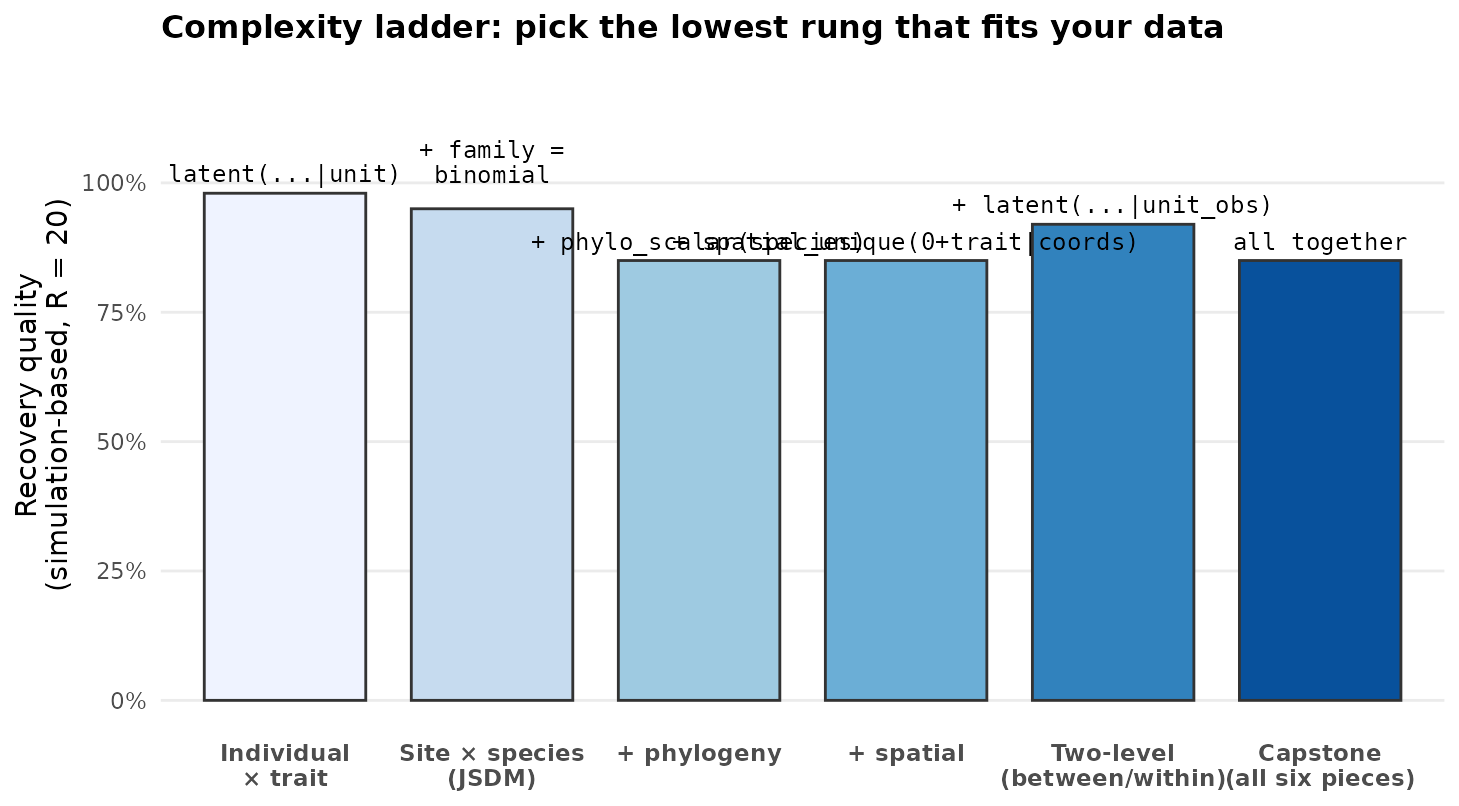
The complexity ladder: each rung adds one structural piece. Bar height = number of identifiable parameter classes recovered cleanly in the simulation-recovery study (Σ_B / Σ_W / sigma2_spa / range / lam_phy). Rungs are stacked left-to-right by complexity; pick the lowest rung that matches your data.
1. What does each row represent?
gllvmTMB is long-format: every fit takes a data frame in
which each row is one (unit, trait) observation, with a
value column for the response and a trait
column naming which trait it is. The first decision is whether your data
have one observational level or
two.
| Your data look like… | Observational levels | Random-effect grouping | Example article |
|---|---|---|---|
| One row per individual / site, with several traits as columns; you reshape to long format. | One: unit
|
latent(0 + trait | unit) |
Morphometrics (individuals × continuous traits) |
| Sites that each contain multiple species, with traits measured for each (site, species) combination; or repeated measures per individual; or papers with multiple items each. | Two: site (between) and site_species
(within) |
latent(0 + trait | site) and latent(0 +
trait | site_species)
|
Corvidae two-stage and Functional biogeography |
If you have a single observational level, the unit
column can be called anything (individual,
site, study, paper); pass its
name to gllvmTMB(..., unit = "your_column"). The two-level
case needs both site and site_species columns;
the simulate_site_trait() helper shows the canonical
layout.
2. What is the response distribution?
Latent structure is shared across response distributions, but the likelihood term per row is family-specific. Pick the family that matches your trait scale.
| Trait values look like… |
family argument |
Notes |
|---|---|---|
| Continuous, real-valued (mass, length, log-abundance) | family = gaussian() |
Identity link. Default. |
| 0/1 presence-absence, yes-no, item-passed | family = binomial() |
Logit link. Used for joint species distribution models and binary-item systematic mapping; see Joint SDM. |
| Counts (offspring, observations) | family = poisson() |
Log link. |
| Different traits use different families |
family = list(gaussian(), binomial(), poisson()) plus a
family_var column |
One family per row. See Response families. |
For the mixed-response case the family list and the
family_var attribute look like this:
fam <- list(gaussian(), binomial(), poisson())
attr(fam, "family_var") <- "family" # column on `data` selecting the row's familyIf you are unsure, start with the family that matches your most abundant trait type and read the response-family reference before committing to a list.
3. Are rows phylogenetically related?
A phylogenetic random effect is appropriate when species (or other taxa) appear repeatedly in your data and closely related taxa are likely to share trait values for evolutionary reasons. The appropriate term depends on the answer to two follow-up questions: do you want a single shared phylogenetic variance per trait, or trait-specific reduced-rank phylogenetic loadings? And how many species do you have?
| Your situation | Add to formula (in-keyword tree = or
vcv =) |
|---|---|
| Rows are not phylogenetically structured (e.g. individuals from one species, or sites without taxonomic identity). | Nothing. |
| Phylogenetic structure with one shared phylogenetic variance per trait. | + phylo_scalar(species, vcv = Cphy) |
Phylogenetic structure with reduced-rank trait-specific loadings (a
phylogenetic version of latent()). |
+ phylo_latent(species, d = K, vcv = Cphy) or
+ phylo_latent(species, d = K, tree = tree)
|
| Many species (n > ~200) and you want speed. | (same phylo_latent term) with tree = tree
(sparse
trick); much faster than vcv = Cphy at this scale. |
The crossover where the sparse
path overtakes the dense path sits around
n_species ≈ 150-200. Above that, the speedup is roughly an
order of magnitude. See Phylogenetic
GLLVM for the benchmark and the choice between
phylo_scalar() and phylo_latent().
4. Are rows spatially organised?
If your sites carry coordinates and you suspect residual spatial autocorrelation, add a sparse SPDE/GMRF random field. Each trait gets its own field by default.
| Your situation | Add to formula | Argument to gllvmTMB()
|
|---|---|---|
| Sites have no spatial structure (or you do not have coordinates). | Nothing. | — |
| Sites have coordinates and each trait should have its own spatial pattern. | + spatial_unique(0 + trait | coords) |
mesh = make_mesh(df, c("lon", "lat"), cutoff = ...) |
| Sites have coordinates and you want one shared spatial-variance scale across traits (parsimony). | + spatial_scalar(0 + trait | coords) |
mesh = make_mesh(df, c("lon", "lat"), cutoff = ...) |
| You want traits to share a low-rank spatial structure. |
+ spatial_latent(0 + trait | coords, d = K) — K shared
SPDE fields driven by a T × K loading matrix Λ_spa. |
mesh = make_mesh(df, c("lon", "lat"), cutoff = ...)
(the spatial analogue of phylo_latent()). |
The SPDE engine wins on speed when the dataset has more than roughly
100 sites; below that, the dense glmmTMB::exp() covariance
is competitive. See SPDE benchmark
for the crossover plot.
5. Do you have a reduced-rank story for trait covariance?
Even with no phylogenetic or spatial structure, you have to decide how the trait covariance itself is parameterised. This is the choice between factor analysis (a few latent dimensions explaining most cross-trait covariance) and unstructured covariance (every pair gets its own parameter).
| Your situation | Add to formula | Notes |
|---|---|---|
| You expect a small number of latent dimensions (size, shape, life-history pace) to explain most of the cross-trait covariance. T traits, K << T factors. | + latent(0 + trait | site, d = K) |
The default GLLVM choice. K = 1–3 is typical. |
| You want full unstructured between-unit covariance with no rank reduction. |
+ unique(0 + trait | site) (specific variance) plus a
separate term for off-diagonal structure as needed |
Reasonable when T is small (T ≤ 5) and you have many units. |
| Confirmatory: you know which traits load on which factor and want to pin those entries of . |
+ latent(0 + trait | site, d = K) plus
lambda_constraint = list(B = M)
|
M is a T × K matrix with NA for free
entries and numeric values for fixed ones. See Lambda constraint;
?suggest_lambda_constraint proposes a sensible matrix from
a fitted unconstrained model. |
For two-level models you typically include both latent()
and unique() at each level, so the full between-unit piece
is
latent(0 + trait | site, d = K) + unique(0 + trait | site)
and the within-unit piece substitutes the within-unit grouping column
(e.g. site_species for site × species data, or the value
passed via unit_obs = "..." for other domains). The
latent() term gives the shared latent factors; the
unique() term gives each trait its own residual variance on
top.
6. Final formula assembly
Below are the canonical formulas for the most common combinations. The chunks below run live to confirm the formulas parse; each fits in a few seconds on the small simulations.
6a. Individuals × continuous traits, no extra structure
The simplest case (rung 0): one level, Gaussian, no phylogeny, no space. See Morphometrics for the full walkthrough.
set.seed(1)
n_ind <- 30; n_traits <- 4
df_ind <- data.frame(
individual = factor(rep(seq_len(n_ind), each = n_traits)),
trait = factor(rep(paste0("trait_", seq_len(n_traits)), n_ind),
levels = paste0("trait_", seq_len(n_traits))),
value = rnorm(n_ind * n_traits)
)
fit_ind <- gllvmTMB(
value ~ 0 + trait + latent(0 + trait | individual, d = 2),
data = df_ind,
unit = "individual"
)6b. Sites × species, presence/absence
Rung 1 with a binomial response: a joint species distribution model. See Joint SDM.
set.seed(2)
sim_b <- simulate_site_trait(
n_sites = 30, n_species = 6, n_traits = 4,
mean_species_per_site = 4,
Lambda_B = matrix(rnorm(8, sd = 0.6), 4, 2),
S_B = rep(0.3, 4), seed = 2
)
df_b <- sim_b$data
df_b$value <- as.integer(df_b$value > 0)
fit_jsdm <- gllvmTMB(
value ~ 0 + trait +
latent(0 + trait | site, d = 2) +
unique(0 + trait | site),
data = df_b,
family = binomial()
)6c. Add phylogeny
Rung 2: keep your favourite formula and add
phylo_scalar() (single shared phylo variance) or
phylo_latent() (reduced-rank). With a tree in hand, prefer
tree = tree (in-keyword) for the sparse
path. See Phylogenetic GLLVM.
set.seed(3)
n_sp <- 8
tree <- ape::rcoal(n_sp); tree$tip.label <- paste0("sp", seq_len(n_sp))
Cphy <- ape::vcv(tree, corr = TRUE)
sim_p <- simulate_site_trait(
n_sites = 30, n_species = n_sp, n_traits = 4,
mean_species_per_site = 5,
Lambda_B = matrix(rnorm(8, sd = 0.5), 4, 2),
S_B = rep(0.3, 4),
Cphy = Cphy, sigma2_phy = rep(0.4, 4),
seed = 3
)
df_p <- sim_p$data
levels(df_p$species) <- tree$tip.label
fit_phy <- gllvmTMB(
value ~ 0 + trait +
latent(0 + trait | site, d = 2) + unique(0 + trait | site) +
phylo_scalar(species, vcv = Cphy),
data = df_p
)6d. Add space
Rung 3: append spatial_unique(0 + trait | coords) and
pass a mesh. The simulator places sites in [0,1]^2
automatically when spatial_range is supplied. See SPDE benchmark.
set.seed(4)
sim_s <- simulate_site_trait(
n_sites = 40, n_species = 1, n_traits = 3,
mean_species_per_site = 1,
Lambda_B = matrix(0.0, 3, 1), S_B = rep(0.1, 3),
spatial_range = 0.3, sigma2_spa = rep(0.4, 3),
seed = 4
)
df_s <- sim_s$data
mesh <- make_mesh(df_s, c("lon", "lat"), cutoff = 0.1)
fit_spa <- gllvmTMB(
value ~ 0 + trait + spatial_unique(0 + trait | coords, mesh = mesh),
data = df_s
)6e. Two-level + phylo + space (capstone)
The full Nakagawa et al. functional-biogeography model: two levels, phylogeny, and space all at once. The formula reads as a sum of the pieces from sections 1–5. The runnable end-to-end version lives in Functional biogeography; in this article we just show the formula:
fit_full <- gllvmTMB(
value ~ 0 + trait + (0 + trait):env_1 + (0 + trait):env_2 +
spatial_unique(0 + trait | coords, mesh = mesh) +
latent(0 + trait | site, d = 2) +
unique(0 + trait | site) +
latent(0 + trait | site_species, d = 1) +
unique(0 + trait | site_species) +
phylo_scalar(species, vcv = Cphy),
data = df
)7. Diagnostic checklist
After any fit, run through these in order:
| Check | How |
|---|---|
| Optimiser convergence and Hessian PD-ness. | sanity_multi(fit) |
| Implied between-unit trait covariance. |
extract_Sigma(fit, level = "unit") (and
extract_Sigma(fit, level = "unit_obs") for two-level
fits). |
| Interpretable loadings. | getLoadings(fit, level = "unit", rotate = "varimax") |
Did you fit unconstrained latent() with
d > 1? Loadings are not unique. |
Use rotate = "varimax" for a quick post-hoc rotation,
or call suggest_lambda_constraint() to propose a
confirmatory lambda_constraint matrix and refit. |
| Communalities / ICCs (manuscript Eqs. 13–15). |
extract_ICC_site(fit),
extract_communality(fit, level = "unit"),
extract_communality(fit, level = "unit_obs")
|
If sanity_multi() flags a non-PD Hessian or a near-zero
loading diagonal, the most common causes are an over-rich model
(d too large, too many latent() terms relative
to data) or factor-level ordering issues in the long-format data. The Morphometrics article has a worked example
of the factor-level pitfall.
See also
- Morphometrics — rung 0: individuals × continuous traits.
- Joint SDM — rung 1: binary multivariate.
- Phylogenetic GLLVM — rung 2: phylogenetic dependence.
- SPDE benchmark — rung 3: spatial dependence.
- Corvidae two-stage — rung 4: two observational levels.
- Functional biogeography — rung 5: capstone.
- Response families — supported likelihoods and mixed-family dispatch.
-
Lambda constraint —
confirmatory factor analysis with fixed loadings; see also
?suggest_lambda_constraint. - Pitfalls — common gotchas in data prep, formula construction, and interpretation.
- Simulation-based recovery — recovery study across the complexity ladder.