Before running any phylogenetic comparative analysis — PGLS, phylogenetic mixed models, ancestral state reconstruction — species names in your data must match the tip labels in your tree. In practice, they rarely do. prepR4pcm automates the matching of species names between data and tree (which we call reconciliation), records every name-matching decision so you can audit it later, and produces an aligned data frame + pruned tree (the aligned objects) where the species lists match exactly — the precondition for any phylogenetic comparative method.
The problem
Mismatches between data and tree arise from three kinds of difference:
-
Formatting differences — same species, written
differently. For example, the same animal may appear as
Homo_sapiensin the tree and asHomo sapiensin the data; trailing whitespace and attached authority strings (Homo sapiens Linnaeus, 1758) cause similar mismatches. - Synonymy — situations where multiple scientific names refer to the same taxonomic group (often a species or genus); for example, when a recent taxonomic revision moved a species to a different genus, the older and newer names both circulate in the literature. See Synonym (taxonomy) on Wikipedia for a fuller introduction.
- Missing names — species in the data but not the tree, or in the tree but not the data, with no naming-rule that would link them.
Fixing these by hand is tedious, error-prone, and poorly documented.
prepR4pcm solves this with a structured matching
cascade of algorithms: exact match → normalised match → synonym
resolution. Every decision is recorded by the software in the
reconciliation result, where you can inspect it via
reconcile_mapping() or
reconcile_summary().
Installation
# Install pak if you don't have it
# install.packages("pak")
# Install prepR4pcm from GitHub
pak::pak("itchyshin/prepR4pcm")Example 1: Reconcile a dataset against a tree
Suppose you have trait data and a phylogenetic tree with slightly different naming conventions.
# Simulated trait data for 6 primate species
trait_data <- data.frame(
species = c(
"Homo sapiens",
"Pan_troglodytes", # underscore instead of space
"Gorilla gorilla",
"Pongo pygmaeus",
"Macaca mulatta",
"Cebus capucinus"
),
body_mass = c(70, 50, 160, 80, 8, 3),
brain_mass = c(1.35, 0.39, 0.50, 0.37, 0.11, 0.07)
)
# Simulated phylogenetic tree (built manually for this example)
tree <- ape::read.tree(text = paste0(
"((((Homo_sapiens:5,Pan_troglodytes:5):3,",
"Gorilla_gorilla:8):4,Pongo_pygmaeus:12):6,",
"(Macaca_mulatta:10,Papio_anubis:10):8);"
))
tree$tip.label # the tip labels (species names) on the tree
#> [1] "Homo_sapiens" "Pan_troglodytes" "Gorilla_gorilla" "Pongo_pygmaeus"
#> [5] "Macaca_mulatta" "Papio_anubis"
plot(tree) # quick visual; underscores in tip labels render as spaces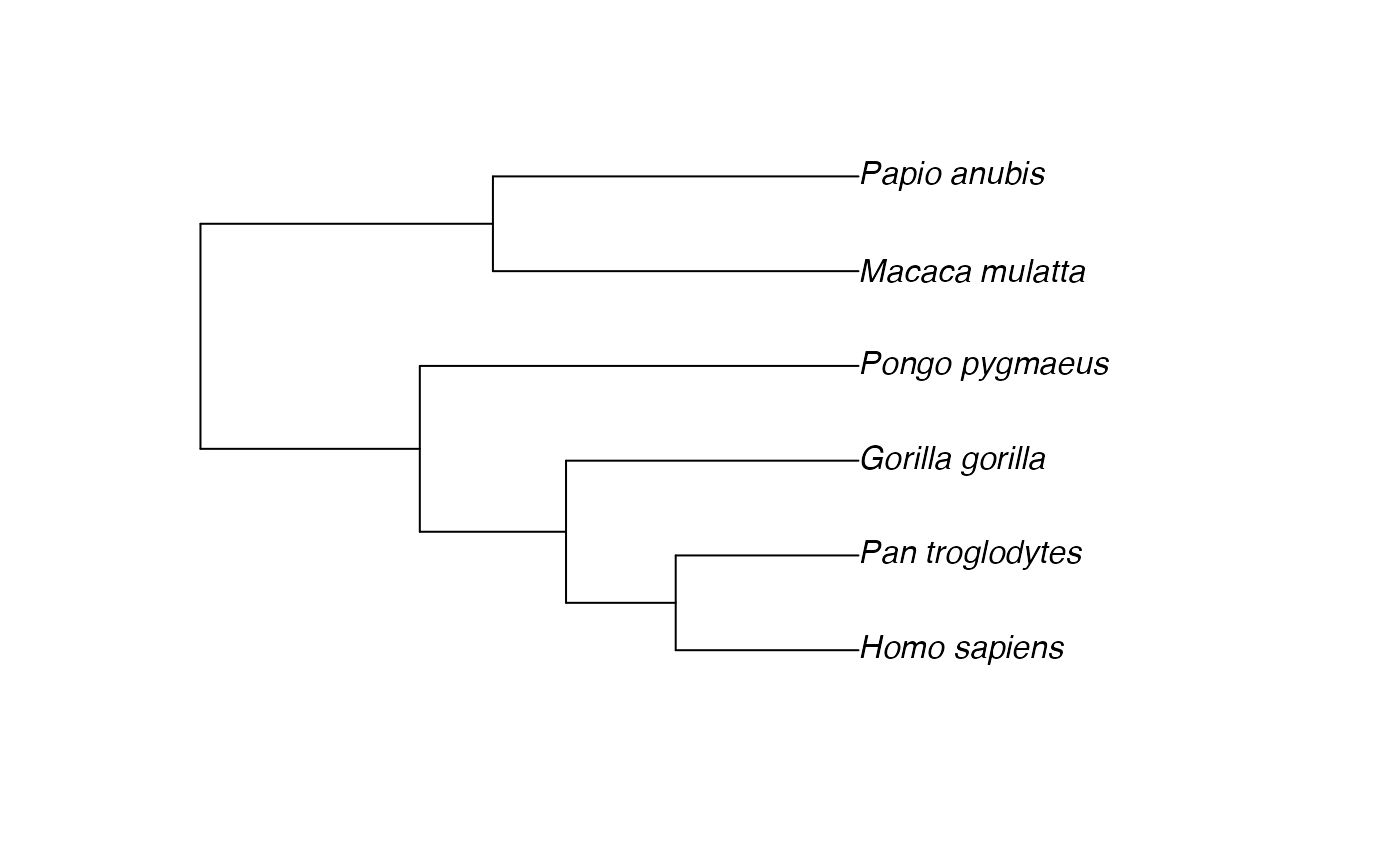
(ape::plot.phylo() displays underscores as spaces by
default — the underlying tree$tip.label strings still
contain underscores, which is why tree$tip.label shows
them.)
Notice the mismatches:
-
Pan_troglodytesin the data has an underscore; the tree uses underscores throughout, but the data column mixes spaces and underscores. -
Cebus capucinusis in the data but not in the tree. -
Papio anubisis in the tree but not in the data.
result <- reconcile_tree(
x = trait_data,
tree = tree,
x_species = "species",
authority = NULL, # skip synonym lookup for this example
quiet = FALSE
)
#> ℹ Reconciling 6 data names vs 6 tree tips
#> ✔ Matched 5/6 data names to tree tipsInspect the result
print(result)
#>
#> ── Reconciliation: data vs tree ────────────────────────────────────────────────
#> Source x: trait_data
#> Source y: phylo (6 tips)
#> Authority: none
#> Timestamp: 2026-06-16 10:10:52
#> ℹ Match coverage: [█████████████████████████░░░░░] 83% (5/6)
#>
#> ── Match summary ──
#>
#> • Exact: 1 (16.7%)
#> • Normalized: 4 (66.7%)
#> • Synonym: 0 ( 0.0%)
#> • Fuzzy: 0 ( 0.0%)
#> • Manual: 0 ( 0.0%)
#> ! Unresolved (x only):1 (16.7%)
#> ! Unresolved (y only):1
#> ! Flagged for review: 0
#> ℹ Use `reconcile_summary()` for details, `reconcile_mapping()` for the full table.The “Reconciliation: data vs tree” header at the top of the output
tells you the call that produced the result; the “Match summary” block
underneath gives the count in each match category (exact, normalised,
synonym, fuzzy, manual, unresolved). Use
reconcile_mapping() to see the full per-name table:
reconcile_mapping(result)
#> # A tibble: 7 × 9
#> name_x name_y name_resolved match_type match_score match_source in_x in_y
#> <chr> <chr> <chr> <chr> <dbl> <chr> <lgl> <lgl>
#> 1 Pan_trog… Pan_t… NA exact 1 exact_string TRUE TRUE
#> 2 Homo sap… Homo_… NA normalized 1 normalisati… TRUE TRUE
#> 3 Gorilla … Goril… NA normalized 1 normalisati… TRUE TRUE
#> 4 Pongo py… Pongo… NA normalized 1 normalisati… TRUE TRUE
#> 5 Macaca m… Macac… NA normalized 1 normalisati… TRUE TRUE
#> 6 Cebus ca… NA NA unresolved NA NA TRUE FALSE
#> 7 NA Papio… NA unresolved NA NA FALSE TRUE
#> # ℹ 1 more variable: notes <chr>What the columns mean:
-
name_x— the species name as it appeared in your data (the argumentxtoreconcile_tree()). -
name_y— the matching tip label on your tree (the argumenttreetoreconcile_tree()), orNAif no match was found. -
name_resolved— the canonical name used when synonym resolution applied (the recognised form per the chosen taxonomic authority).NAfor matches that didn’t go through the synonym stage. -
match_type— which stage of the cascade matched the name (see Understanding match types below). -
match_score— confidence on[0, 1](1for exact / normalised / synonym / manual;< 1for fuzzy / flagged). -
in_x,in_y— logical: was this name in the data, in the tree, or both? -
notes— human-readable note (e.g. “normalised: lowercased”, “via synonym lookup against COL”, “fuzzy match score 0.92”).
For a detailed report:
reconcile_summary(result)Apply manual overrides
Suppose you know that Cebus capucinus should not be in
the analysis. You can document this decision:
result <- reconcile_override(
result,
name_x = "Cebus capucinus",
name_y = NA,
action = "reject",
note = "Not in target phylogeny; exclude from analysis"
)
#> ✔ Override applied: 'Cebus capucinus' -> 'NA' (reject)reconcile_override() updates the existing
result (the reconciliation you built earlier)
in place — no need to re-run reconcile_tree(). The three
actions you can pass to action = ... are:
-
"accept"— confirm a specificname_x → name_ymapping. -
"reject"— mark a name as deliberately excluded. -
"replace"— redirectname_xto a differentname_ythan the cascade produced.
Produce aligned objects
Once satisfied with the reconciliation, apply it:
aligned <- reconcile_apply(
result,
data = trait_data,
tree = tree,
species_col = "species",
drop_unresolved = TRUE
)
#> ! Dropped 1 rows with unresolved species from data
#> ℹ Tree has 5 tips after alignment
# Aligned data frame — only species present in both data and tree
aligned$data
#> species body_mass brain_mass
#> 1 Homo sapiens 70 1.35
#> 2 Pan_troglodytes 50 0.39
#> 3 Gorilla gorilla 160 0.50
#> 4 Pongo pygmaeus 80 0.37
#> 5 Macaca mulatta 8 0.11
# Aligned tree — pruned to matched species
ape::Ntip(aligned$tree)
#> [1] 5
plot(aligned$tree) # the pruned tree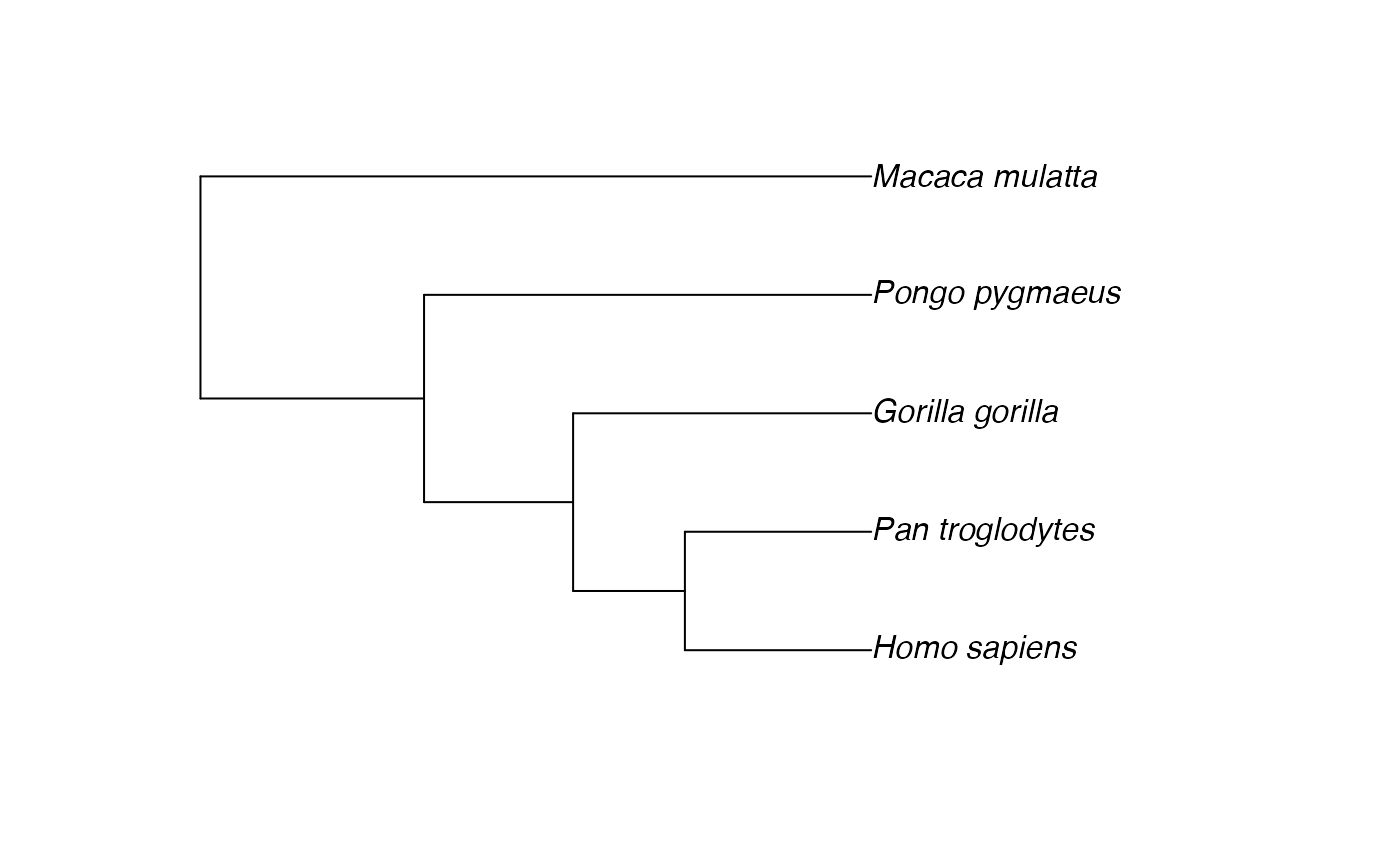
The $data and $tree components now have
matching species, ready for comparative analysis.
Example 2: Reconcile two datasets
prepR4pcm can also reconcile species names between
two datasets, not just between a dataset and a tree. The same
matching cascade applies. This is useful when merging trait data from
different sources, where species names often disagree across datasets.
Here is a toy example:
# df1: body mass for three primates (df1 uses an underscore for chimp)
df1 <- data.frame(
species = c("Homo sapiens", "Pan_troglodytes", "Gorilla gorilla"),
mass = c(70, 50, 160)
)
# df2: lifespan for three primates (df2 uses a space for chimp; orang
# is here but not gorilla)
df2 <- data.frame(
species = c("Homo sapiens", "Pan troglodytes", "Pongo pygmaeus"),
lifespan = c(79, 40, 45)
)
# Reconcile the species columns of df1 and df2 against each other.
# `authority = NULL` skips the synonym-lookup stage (no taxonomic
# database needed for this small example). `quiet = TRUE` suppresses
# progress messages.
result2 <- reconcile_data(
x = df1,
y = df2,
authority = NULL,
quiet = TRUE
)
#> ℹ Auto-detected species column: species
#> ℹ Auto-detected species column: species
# The output shows how many names matched, and via which stage.
print(result2)
#>
#> ── Reconciliation: data vs data ────────────────────────────────────────────────
#> Source x: df1
#> Source y: df2
#> Authority: none
#> Timestamp: 2026-06-16 10:10:52
#> ℹ Match coverage: [████████████████████░░░░░░░░░░] 67% (2/3)
#>
#> ── Match summary ──
#>
#> • Exact: 1 (33.3%)
#> • Normalized: 1 (33.3%)
#> • Synonym: 0 ( 0.0%)
#> • Fuzzy: 0 ( 0.0%)
#> • Manual: 0 ( 0.0%)
#> ! Unresolved (x only):1 (33.3%)
#> ! Unresolved (y only):1
#> ! Flagged for review: 0
#> ℹ Use `reconcile_summary()` for details, `reconcile_mapping()` for the full table.Pan_troglodytes (underscore) in df1 is
matched to Pan troglodytes (space) in df2 via
normalisation. Gorilla gorilla is in df1 only
and Pongo pygmaeus is in df2 only — both end
up as unresolved rows
(in_x = TRUE, in_y = FALSE and vice versa).
Understanding match types
Every row in the reconcile_mapping() output has a
match_type column. Here is what each value means and what
action (if any) it requires:
match_type |
Meaning | Action needed? |
|---|---|---|
exact |
Verbatim string equality | None |
normalized |
Names matched after stripping underscores, authority strings, and case differences | None — check the notes column if you want to
confirm |
synonym |
Names resolved through a taxonomic authority (e.g., Catalogue of Life) to the same accepted name | Verify the resolved name looks correct |
fuzzy |
High-confidence character-level match (score ≥
flag_threshold, default 0.95) |
Check the match_score column; review with
reconcile_suggest()
|
flagged |
Lower-confidence match that needs human review: fuzzy score below
flag_threshold, or an indirect synonym chain |
Review with reconcile_review() or
reconcile_suggest()
|
manual |
Set by reconcile_override() or the
overrides argument |
None — you decided this |
unresolved |
No match found after all stages | Investigate; use reconcile_suggest() for candidates or
reconcile_override() to document a decision |
Use
reconcile_summary(result, detail = "mismatches_only") to
see only the rows that need attention.
Example 3: Using a taxonomic authority
A taxonomic authority is a curated database of species names that records, for each name, which is the currently-recognised one and which are synonyms (alternative names referring to the same taxon). prepR4pcm can use such an authority to recognise that two syntactically different names refer to the same species — the “synonym” stage of the matching cascade.
Most authorities below are databases served by the taxadb package
(Norman et al. 2020) — authority = "col" tells
prepR4pcm to look up synonyms in the taxadb-cached copy of
the Catalogue of Life, and so on. The first call for a taxadb provider
downloads its database to your local cache (~100 MB); subsequent calls
are fast and work offline. One alternative, "gnverifier",
is HTTP-backed instead of taxadb: it calls the Global Names verifier on
each lookup. No database to download, but each lookup needs network
access and the package.
-
col— Catalogue of Life (catalogueoflife.org). Broad coverage; the most general default for cross-taxon work. -
itis— Integrated Taxonomic Information System (itis.gov). North-American emphasis, strong on vertebrates and vascular plants. -
gbif— Global Biodiversity Information Facility taxonomic backbone (datasetd7dddbf4-2cf0-4f39-9b2a-bb099caae36c). Pragmatic synthesis with very wide coverage. -
ncbi— NCBI Taxonomy (ncbi.nlm.nih.gov/taxonomy). Tracks names that appear in GenBank — most useful for molecular-data workflows. -
ott— Open Tree Taxonomy (tree.opentreeoflife.org/about/taxonomy-version). Note:otthere is a taxadb authority name, not an R package. The R package that retrieves trees from Open Tree of Life is calledrotl, which is separate. Useauthority = "ott"if you also usepr_get_tree(source = "rotl")and want the synonym-resolution step to use the same taxonomy as the tree. -
itis_test— small bundled subset of ITIS used for the package’s own examples and tests; not a general-purpose authority. -
gnverifier— Global Names verifier (verifier.globalnames.org). Verifies names against ~100 authoritative sources (CoL, ITIS, GBIF, NCBI, Open Tree, …) in one HTTP call. Wider source coverage than any single taxadb provider and no ~100 MB local download, but each call needs network access and the package.
The taxadb-backed entries mirror the providers documented in
?taxadb::td_create.
When should you set authority?
Use authority = NULL (skip synonym lookup) when:
- You want a quick offline check — no database download required.
- Species names in your data and tree are unlikely to differ much (most formatting differences are caught by the normalisation stage anyway).
Set authority = "col" (or another taxadb provider) when
names differ because of genuine taxonomic revisions — species moved to a
different genus, splits, or lumps. The first run downloads a local
database (~100 MB); subsequent runs are fast because the database is
cached.
Use authority = "gnverifier" when you would rather query
the Global Names verifier over HTTP than maintain a local taxadb
database. It is the right pick when you want broader source coverage
than any one taxadb provider (it consults ~100 sources per call), when
you do not want to download a ~100 MB cache, or when you would like the
synonym stage to silently benefit from upstream-source improvements
without re-downloading anything. The trade-off: every call needs network
access (we degrade to “name not found” on failure, so the rest of the
cascade still runs), and the request adds a round-trip to
verifier.globalnames.org. Install
(install.packages("httr2")) before first use.
# Requires taxadb and a local database download (automatic on first use)
result3 <- reconcile_tree(
x = trait_data,
tree = tree,
x_species = "species",
authority = "col" # Catalogue of Life
)Example 4: Pre-built overrides
Researchers often maintain a curated list of known corrections. You can pass these as a data frame, or as a path to a file in CSV format:
The chunks below use
my_dataandmy_treeas hypothetical objects (substitute your own data frame andphyloobject). They are markedeval = FALSEso the vignette renders without requiring those objects to exist.
# A data frame of known corrections
corrections <- data.frame(
name_x = c("Corvus sp.", "Turdus merulaa"),
name_y = c("Corvus corax", "Turdus merula"),
user_note = c("Only one Corvus in our tree", "Typo in source data")
)
result4 <- reconcile_tree(
x = my_data,
tree = my_tree,
overrides = corrections
)
# Or from a CSV file:
result5 <- reconcile_tree(
x = my_data,
tree = my_tree,
overrides = "lab_corrections.csv"
)Overrides are applied before any other matching stage, so they always take priority.
Example 5: Multiple datasets against one tree
reconcile_multi() reconciles several datasets at once,
pooling all unique species names before running the cascade:
# Suppose you have several data frames to reconcile against one tree.
# `my_ecology_data`, `my_morpho_data`, and `my_tree` are **hypothetical**
# user-supplied objects; substitute your own.
datasets <- list(
traits = trait_data, # defined above
ecology = my_ecology_data, # your own data frame
morpho = my_morpho_data # your own data frame
)
result6 <- reconcile_multi(datasets, my_tree)
print(result6)Key design principles
- Conservative: Names are never silently changed. Ambiguous cases are flagged, not auto-resolved.
- Transparent: Every decision is recorded with match type, score, source, and a human-readable note.
- Reproducible: Database versions are pinned. All parameters used to build the result are stored on the result object itself, so a collaborator can re-run the same reconciliation later.
-
Practical: Works with the data types comparative
biologists already use — a
data.frameof trait values (one row per species) and a phylogenetic tree as anape::phyloobject.
Typical workflow
The chunk below uses hypothetical files (
species_traits.csv,species_tree.nwk) — substitute your own paths. The chunk is markedeval = FALSEso it doesn’t try to read files that don’t exist when the vignette is rendered.
library(prepR4pcm)
# 1. Load your data and tree (hypothetical paths -- substitute your own)
my_data <- read.csv("species_traits.csv")
my_tree <- ape::read.tree("species_tree.nwk")
# 2. Reconcile
result <- reconcile_tree(my_data, my_tree, authority = "col")
# 3. Review
print(result)
reconcile_summary(result, detail = "mismatches_only")
# 4. Fix manually if needed
result <- reconcile_override(result, "Corvus sp.", "Corvus corax",
note = "Only one Corvus in tree")
# 5. Apply
aligned <- reconcile_apply(result, data = my_data, tree = my_tree,
drop_unresolved = TRUE)
# 6. Analyse
# aligned$data and aligned$tree are ready for caper, phytools, MCMCglmm, etc.References
- Hadfield, J.D. (2010) MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R package. Journal of Statistical Software 33:1–22. DOI 10.18637/jss.v033.i02
- Orme, D., Freckleton, R., Thomas, G., Petzoldt, T., Fritz, S., Isaac, N. & Pearse, W. (2025) caper: Comparative Analyses of Phylogenetics and Evolution in R. R package version 1.0.4. DOI 10.32614/CRAN.package.caper
- Paradis, E. & Schliep, K. (2019) ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35:526–528. DOI 10.1093/bioinformatics/bty633
- Revell, L.J. (2024) phytools 2.0: an updated R ecosystem for phylogenetic comparative methods (and other things). PeerJ 12:e16505. DOI 10.7717/peerj.16505